สังเกตได้ในทุก ๆ 68, 000-88,000 ทารกแรกเกิด กลุ่มอาการ Apert เกิดจากการกลายพันธุ์เฉพาะของยีน FGFR2 ซึ่งมีหน้าที่ควบคุมการหลอมละลายของกะโหลกศีรษะและการพัฒนาของนิ้วมือและนิ้วเท้า
สำหรับการวินิจฉัยโรค Apert การตรวจร่างกาย ความทรงจำ การประเมินทางรังสีของกะโหลกศีรษะ นิ้วมือ และนิ้วเท้า และสุดท้าย การทดสอบทางพันธุกรรมเป็นพื้นฐาน
ปัจจุบันผู้ที่เป็นโรค Apert สามารถพึ่งพาการรักษาตามอาการเท่านั้น กล่าวคือ บรรเทาอาการและหลีกเลี่ยงภาวะแทรกซ้อนที่ร้ายแรงที่สุด
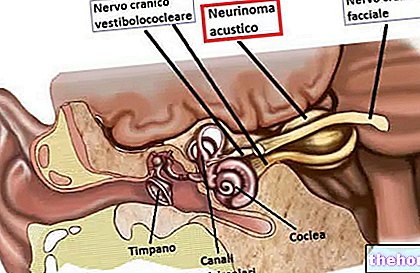
ทบทวนโดยสังเขปของการเย็บกะโหลกและการหลอมรวม
รอยเย็บกะโหลกเป็นข้อต่อที่มีเส้นใย ซึ่งทำหน้าที่หลอมรวมกระดูกของหลุมฝังศพกะโหลกเข้าด้วยกัน (เช่น กระดูกหน้าผาก ขมับ ข้างขม่อม และท้ายทอย)
ภายใต้สภาวะปกติ กระบวนการหลอมรวมของการเย็บกะโหลกศีรษะจะเกิดขึ้นในช่วงหลังคลอด โดยเริ่มตั้งแต่อายุ 1-2 ปี สำหรับองค์ประกอบร่วมบางอย่าง และสิ้นสุดที่อายุ 20 สำหรับคนอื่นๆ กระบวนการหลอมรวมที่ยาวและต่อเนื่องช่วยให้สมองเติบโตและพัฒนาได้อย่างเพียงพอ
อย่างไรก็ตาม โรค Apert เป็นหนี้ความอื้อฉาวไม่เพียง แต่เกี่ยวข้องกับโรคกะโหลกศีรษะ แต่ยังรวมถึงความจริงที่ว่ามันเกี่ยวข้องกับระดับของ syndactyly นั่นคือความผิดปกติ แต่กำเนิดที่โดดเด่นด้วยการรวมนิ้วหรือนิ้วหนึ่งนิ้วหรือมากกว่า เท้า.
ความเป็นไปได้ที่จะทำให้เกิด craniostenosis และ syndactyly ในเวลาเดียวกันทำให้ Apert's syndrome เป็นตัวอย่างของ acrocephalosyndactyly ในทางการแพทย์ "acrocephalosyndactyly เป็นภาวะทางพันธุกรรมที่รวมความผิดปกติเฉพาะของกะโหลกศีรษะ (" acrocephalus "หมายถึง" หัวถึงปลาย ") กับการหลอมรวมของนิ้วหรือนิ้วเท้าหนึ่งนิ้วหรือมากกว่า
อะไรคือผลที่ตามมาของการเชื่อมประสานกะโหลกในระยะแรก?
เช่นเดียวกับในกรณีของโรค Apert และโรคอื่นๆ ที่เกี่ยวข้อง การหลอมรวมของรอยเย็บกะโหลกเกิดขึ้นระหว่างก่อนคลอด ปริกำเนิด (*) หรือวัยเด็กตอนต้น อวัยวะของสมอง เช่น สมอง สมองน้อย และก้านสมอง และประสาทสัมผัสเมื่อได้รับสายตา การเปลี่ยนแปลงทั้งการเจริญเติบโตและรูปร่าง
* หมายเหตุ: "ชีวิตปริกำเนิด" หมายถึงช่วงเวลาระหว่างสัปดาห์ที่ 27 ของการตั้งครรภ์และ 28 วันแรกหลังคลอด
ระบาดวิทยา: Apert Syndrome เป็นอย่างไร?
จากสถิติพบว่า 1 ใน 65,000-88,000 คนเกิดมาพร้อมกับโรค Apert
คุณรู้หรือเปล่าว่า ...
โรคทางพันธุกรรมเช่น Apert syndrome ทำให้เกิด craniosynostosis ประมาณ 150
นอกเหนือจากกลุ่มอาการ Apert แล้ว Crouzon syndrome, Pfeiffer syndrome และ Saethre-Chotzen syndrome ยังมีความสำคัญ
ความอยากรู้
การกลายพันธุ์ที่ได้มาซึ่งทำให้เกิดกลุ่มอาการ Apert เป็นตัวอย่างของ "การกลายพันธุ์ เดอโนโว"นั่นคือของ" การกลายพันธุ์ใหม่อย่างสมบูรณ์ปราศจากลักษณะทางพันธุกรรม "
อะไรทำให้เกิดการกลายพันธุ์ของยีนที่เกี่ยวข้องกับกลุ่มอาการ Apert?
ที่ตั้ง: ยีนที่มีอยู่ในโครโมโซมของมนุษย์เป็นลำดับดีเอ็นเอที่มีหน้าที่ในการผลิตโปรตีนพื้นฐานในกระบวนการทางชีววิทยาที่จำเป็นต่อชีวิต รวมทั้งการเจริญเติบโตของเซลล์และการจำลองแบบ
เมื่อไม่มีการกลายพันธุ์ (เพราะฉะนั้นในคนที่มีสุขภาพดี) ยีน FGFR2 ที่เกี่ยวข้องกับกลุ่มอาการ Apert จะผลิตโปรตีนตัวรับในปริมาณที่เหมาะสม ซึ่งเรียกว่า Fibroblast Growth Factor Receptor 2 ซึ่งจำเป็นต่อการทำเครื่องหมายเวลาของการหลอมรวมของกะโหลกศีรษะ เย็บและเพื่อตรวจสอบการแยกนิ้วและนิ้วเท้า (กล่าวอีกนัยหนึ่งคือสัญญาณเมื่อถึงเวลาที่เหมาะสมสำหรับการหลอมรวมของเย็บกะโหลกและควบคุมการก่อตัวของนิ้วมือและนิ้วเท้า)
ในทางกลับกัน เมื่อผ่านการกลายพันธุ์ที่สังเกตพบในการปรากฏตัวของกลุ่มอาการ Apert ยีน FGFR2 จะกระทำมากกว่าปกและผลิตโปรตีนตัวรับดังกล่าวในปริมาณมหาศาล ซึ่งระยะเวลาที่เกี่ยวข้องกับการหลอมรวมของรอยเย็บกะโหลกจะเปลี่ยนไป (มันคือ เร็วขึ้น) และกระบวนการแยกนิ้วและนิ้วเท้าไม่เกิดขึ้นอย่างถูกต้อง
ใครมีความเสี่ยงมากที่สุด?
เกี่ยวกับกรณีที่ได้รับของ Apert syndrome ปัจจัยที่กระตุ้นการกลายพันธุ์ของยีน FGFR2 หลังจากการปฏิสนธิยังไม่ชัดเจนในขณะนี้
การวิจัยในด้านนี้ยังคงดำเนินต่อไป
Apert syndrome เป็นโรคที่มีลักษณะเด่นของ autosomal
เข้าใจไหม...
ยีนมนุษย์แต่ละยีนมีอยู่ในสองสำเนา เรียกว่าอัลลีล ยีนหนึ่งมาจากมารดาและอีกอันหนึ่งมาจากบิดา
Apert syndrome มีลักษณะเฉพาะของโรคที่โดดเด่น autosomal
โรคทางพันธุกรรมเป็นลักษณะเด่นของ autosomal เมื่อการกลายพันธุ์ของยีนเดียวที่ทำให้เกิดการกลายพันธุ์นั้นเพียงพอที่จะแสดงออก
- ใบหน้าแบนหรือเว้า (เนื่องจากการเติบโตของกระดูกกลางของใบหน้าไม่เพียงพอ)
- บวม โป่ง และตาเบิกกว้าง เบ้าตาตื้นและดวงตาเว้นระยะผิดปกติ (hypertelorism ของเบ้าตา);
- จมูกจงอย;
- กรามด้อยพัฒนารวมกับการพยากรณ์โรค
- ฟันซ้อน (เนื่องจากกรามด้อยพัฒนา)
- หูชั้นต่ำกว่าปกติ
Syndactyly
ในพาหะของโรค Apert มักพบเห็น syndactyly ในมือ เกือบตลอดเวลา และที่เท้า น้อยกว่าในมือ

ลักษณะทั่วไปของ syndactyly ในมือของบุคคลที่มีอาการ Apert คือ 4:
- การปรากฏตัวของนิ้วหัวแม่มือสั้นที่มีการเบี่ยงเบนในแนวรัศมี (กล่าวคือ ชี้ไปทางรัศมีอย่างผิดปกติ หนึ่งในสองกระดูกของปลายแขน);
- ซับซ้อนระหว่างนิ้วชี้ นิ้วกลาง และนิ้วนาง โดยความซับซ้อน syndactyly แพทย์หมายถึงการหลอมรวมของนิ้วมือที่ผิดปกติซึ่งส่งผลต่อเนื้อเยื่ออ่อน (ผิวหนัง) ไม่เพียงเท่านั้น แต่ยังรวมถึงเนื้อเยื่อกระดูก (phalanges) ด้วย
- การแสดงความเห็นอกเห็นใจ เป็นศัพท์ทางการแพทย์ที่บ่งบอกถึงการหลอมรวมที่ผิดปกติของข้อต่อระหว่างกระดูกของนิ้ว
- syndactyly ง่าย ๆ ระหว่างนิ้วเท้าที่ 4 และ 5 (เช่น ระหว่างนิ้วนางกับนิ้วก้อย) ด้วย Syndactyly ง่าย ๆ ผู้เชี่ยวชาญกล่าวถึงการประสานนิ้วที่ผิดปกติซึ่งส่งผลต่อเนื้อเยื่ออ่อน (ผิวหนัง) เท่านั้น
ความรุนแรงของโรคในกลุ่มอาการเปิด 3 ประเภท
ตามความรุนแรงของอาการนิ้วหัวแม่มือผิดรูป (ลักษณะแรกจากสี่ลักษณะ) ผู้เชี่ยวชาญกลุ่มอาการ Apert แยกแยะกลุ่มอาการของความรุนแรงที่เพิ่มขึ้น 3 ประเภท:
- ประเภทที่ 1 (รุนแรงน้อยที่สุด) เกิดขึ้นพร้อมกับ "ความผิดปกติน้อยที่สุดที่ส่งผลต่อนิ้วหัวแม่มือ ซึ่งยังคงเป็นอิสระจากดัชนีโดยสิ้นเชิง"
ความผิดปกติอื่นๆ: นิ้วชี้ นิ้วกลาง และนิ้วนางหลอมรวมกันผ่านซินแด็กทีลีและซิมโฟแลงซิสที่ซับซ้อนซึ่งส่งผลต่อข้อต่อส่วนปลาย c "เรียบง่ายและไม่สมบูรณ์ syndactyly ระหว่างแหวนและนิ้วก้อย (syndactyly ที่ไม่สมบูรณ์หมายความว่าฟิวชั่นระหว่างสองนิ้วเป็นบางส่วน)
ข้อมูลอื่น ๆ: เป็นประเภทที่พบบ่อยที่สุด - ประเภท II (ความรุนแรงระดับกลาง) มีลักษณะเฉพาะโดยมีการเบี่ยงเบนในแนวรัศมีของนิ้วโป้งมากกว่า เมื่อเทียบกับกรณีก่อนหน้า และโดยหลักการประสานกันระหว่างนิ้วโป้งกับนิ้วชี้ (c "คือความสัมพันธ์ที่ไม่สมบูรณ์ระหว่างนิ้วโป้งกับนิ้วชี้) .
ความผิดปกติอื่นๆ: นิ้วชี้ นิ้วกลาง และนิ้วนางเป็นตัวชูโรงของซินแด็กทีลีที่รวมเข้ากับการแสดงความเห็นอกเห็นใจส่วนปลาย ระหว่างนิ้วนางกับนิ้วก้อย ค "เป็นความสัมพันธ์ที่เรียบง่ายและไม่สมบูรณ์
ข้อมูลอื่น ๆ: เป็นประเภทที่พบมากเป็นอันดับสอง - ประเภทที่ 3 (รุนแรงที่สุด) มีลักษณะเป็นนิ้วโป้งที่ต่อเข้ากับดัชนี ไม่เพียงแต่ในระดับเนื้อเยื่ออ่อนเท่านั้น แต่ยังรวมถึงระดับของเนื้อเยื่อกระดูกด้วย
ความผิดปกติอื่นๆ: นิ้วทั้งหมดหลอมรวมกันจนแทบจะจำไม่ได้ c "เป็น" เล็บเดียว; ถ้าระหว่าง 4 นิ้วแรก syndactyly นั้นซับซ้อน ระหว่างนิ้วนางกับนิ้วก้อยนั้น (สำหรับประเภทอื่นๆ) นั้นเรียบง่ายและไม่สมบูรณ์
ข้อมูลอื่น ๆ: เป็นประเภทที่หายากที่สุด
อาการและอาการแสดงอื่นๆ ที่เป็นไปได้ของโรค Apert
ในบางกรณี นอกเหนือจากความเกี่ยวข้องกับ craniosynostosis และ syndactyly แล้ว กลุ่มอาการ Apert ยังสัมพันธ์กับการมีอยู่ของ: polydactyly (เช่น มีนิ้วพิเศษอยู่ในมือหรือเท้า) การสูญเสียการได้ยิน หูและไซนัสกำเริบ เหงื่อออกมาก ผิวมัน ผิวหนัง สิวรุนแรง ขนคิ้วไม่ติด กระดูกสันหลังส่วนคอ ภาวะหยุดหายใจขณะหลับอุดกั้น และ/หรือเพดานโหว่
ภาวะแทรกซ้อน
ภาวะแทรกซ้อนของ Apert syndrome เหนือสิ่งอื่นใดคือผลกระทบร้ายแรงที่ craniosynostosis สามารถมีต่อการพัฒนาสมองและความสามารถทางปัญญาและความสามารถในการทำงานของมือขึ้นอยู่กับ syndactyly
สามารถตรวจพบกลุ่มอาการ Apert ได้เมื่อใด
โดยปกติ ความผิดปกติของกะโหลกศีรษะและดิจิทัลอันเนื่องมาจากกลุ่มอาการ Apert จะปรากฏชัดตั้งแต่แรกเกิด ดังนั้นการวินิจฉัยและการวางแผนการรักษาจึงเกิดขึ้นได้ในทันที
ไปที่ศีรษะ (เอ็กซ์เรย์ของศีรษะ, CT scan ของศีรษะและ / หรือ MRI ของศีรษะ) และของมือและเท้า; ในที่สุดก็จบลงด้วยการทดสอบทางพันธุกรรม
การตรวจร่างกายและประวัติการรักษา
การตรวจร่างกายและประวัติโดยพื้นฐานแล้วประกอบด้วยการตรวจสอบอาการที่แสดงโดยผู้ป่วยอย่างแม่นยำ
ในบริบทของ Apert syndrome ในช่วงเวลาเหล่านี้ของกระบวนการวินิจฉัยที่แพทย์พบ craniosynostosis และ syndactyly และลักษณะที่แม่นยำของพวกเขา
การตรวจทางรังสีของศีรษะ นิ้ว และนิ้วเท้า
ในบริบทของโรค Apert:
- แพทย์ใช้การตรวจทางรังสีของศีรษะเพื่อยืนยันการปรากฏตัวของการหลอมรวมของรอยประสานของหลอดเลือด (craniosynostosis coronal craniosynostosis หรือ brachycephaly); นอกจากนี้ยังช่วยให้เขาประเมินความรุนแรงของความผิดปกติของกะโหลกศีรษะและสมองในปัจจุบันได้
- ในทางกลับกัน การตรวจทางรังสีของนิ้วมือและนิ้วเท้านั้นไม่จำเป็นมากนักสำหรับการยืนยัน syndactyly (สำหรับสิ่งนี้ การตรวจด้วยสายตาก็เพียงพอแล้ว) แต่หากต้องการทราบรายละเอียดเกี่ยวกับลักษณะของฟิวชั่นระหว่างดิจิทัล (ประเภทของ syndactyly ปัจจุบัน ระดับ ของฟิวชั่น เป็นต้น)
การทดสอบทางพันธุกรรม
เป็นการวิเคราะห์ดีเอ็นเอที่มุ่งตรวจหาการกลายพันธุ์ในยีนที่สำคัญ
ในบริบทของ Apert syndrome การทดสอบนี้แสดงถึงการทดสอบวินิจฉัยยืนยัน เนื่องจากทำให้เห็นลักษณะการกลายพันธุ์ของ FGFR2 ของโรคทางพันธุกรรมที่เป็นปัญหา
การดูแลศัลยกรรมของ BRACHYCEPHALIA
สำหรับพาหะของ Apert syndrome การผ่าตัดรักษา brachycephaly รวมถึง:
- การแทรกแซงครั้งแรกตั้งแต่อายุยังน้อย (ภายในหนึ่งปีแห่งชีวิต) โดยมุ่งเป้าไปที่การแยกรอยประสานรอยต่อของหลอดเลือดหัวใจก่อนเวลาที่คาดไว้ หากการแทรกแซงนี้ประสบผลสำเร็จ สมองจะได้รับพื้นที่ที่เหมาะสมสำหรับการเจริญเติบโตและลดความเสี่ยงของปัญหาทางปัญญา
- การแทรกแซงครั้งที่สองระหว่างอายุ 4 ถึง 12 ปีโดยมุ่งเป้าไปที่การทำให้ใบหน้าดูเป็นปกติ ซึ่ง (ตามที่ผู้อ่านจะจำได้) จะแบนหากไม่เว้า
การผ่าตัดที่เป็นปัญหาเกี่ยวข้องกับการกรีดกระดูกใบหน้าบางส่วนและการจัดตำแหน่งตามการจัดเรียงที่สะท้อนถึงความปกติอย่างน้อยบางส่วน - การแทรกแซงในที่สุดครั้งที่สามในช่วงหลายปีของวัยเด็กโดยมีจุดประสงค์เพื่อกำจัดหรืออย่างน้อยก็เพื่อลดการเกิด hypertelorism ในตา
การดูแลศัลยกรรมของซินดาซี
การผ่าตัดรักษา syndactyly แตกต่างกันไปตามลักษณะของฟิวชั่น interdigital (จึงขึ้นอยู่กับประเภท)

ซึ่งหมายความว่าการแทรกแซงที่ถูกต้องสำหรับบุคคลที่มีอาการ Apert อาจไม่ถูกต้องสำหรับบุคคลอื่นที่มีโรคทางพันธุกรรมเดียวกัน (จะมีผลก็ต่อเมื่อชนิดของ syndactyly มีอยู่เหมือนกัน)
เมื่อชี้แจงประเด็นพื้นฐานนี้แล้ว เป้าหมายของวิธีการผ่าตัดที่มีอยู่แต่ละประเภทก็เหมือนกันและประกอบด้วยการปล่อยนิ้วที่หลอมละลายเพื่อรับประกันการทำงานบางอย่างของมือ
โดยทั่วไป การรักษา syndactyly เกี่ยวข้องกับสองขั้นตอน:
- 1 ขั้นตอน: "ว่าง" ช่องว่างระหว่างนิ้วแรก (ช่องว่างระหว่างนิ้วโป้งกับนิ้วชี้) และช่องว่างระหว่างนิ้วที่สี่ (ช่องว่างระหว่างนิ้วนางกับนิ้วก้อย);
- 2 ขั้นตอน: "ว่าง" ช่องว่างระหว่างนิ้วที่สองและสาม (ช่องว่างระหว่างนิ้วชี้และนิ้วกลาง และช่องว่างระหว่างนิ้วกลางและนิ้วนาง)









---pan-di-spagna-per-rotolo-dolce.jpg)