ลักษณะทั่วไป
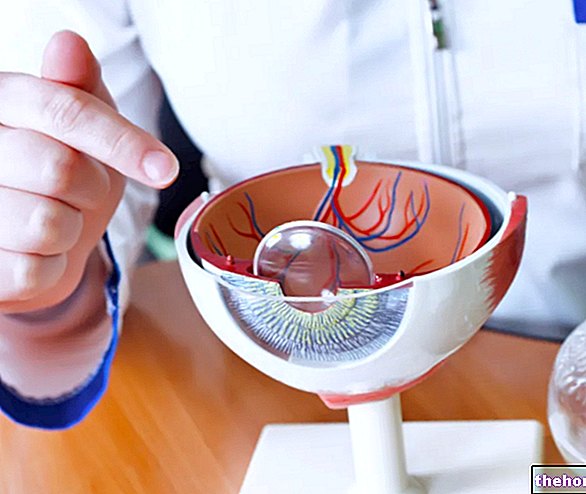
Retinoblastoma (Rb) เป็นเนื้องอกมะเร็งที่พัฒนาจากเซลล์ของเรตินา มะเร็งนี้สามารถเกิดขึ้นได้ทุกเพศทุกวัย แต่การเริ่มมีอาการมักเกิดขึ้นในช่วงวัยทารกก่อนอายุห้าขวบ

มะเร็งในวัยเด็กมีความก้าวร้าว: มะเร็งจอประสาทตาสามารถแพร่กระจายไปยังต่อมน้ำเหลือง กระดูก หรือไขกระดูก ไม่ค่อยเกี่ยวข้องกับระบบประสาทส่วนกลาง (สมองและไขสันหลัง)
เด็กประมาณ 90% ที่เป็นโรคเรติโนบลาสโตมามีการพยากรณ์โรคในเชิงบวก (ความน่าจะเป็นของการรักษา) โดยที่การวินิจฉัยต้องเร็วและต้องเริ่มการรักษาก่อนที่มะเร็งจะลุกลาม เมื่อใดก็ตามที่เป็นไปได้ เป้าหมายของการแทรกแซงทางการแพทย์คือการรักษาวิสัยทัศน์ของผู้ป่วย
สาเหตุ
ชุดของเหตุการณ์ที่นำไปสู่การโจมตีของเนื้องอกนั้นซับซ้อน สิ่งนี้เริ่มต้นเมื่อเซลล์ในเรตินาพัฒนาการกลายพันธุ์ (หรือการลบออก) ที่เกี่ยวข้องกับยีนต้านเนื้องอก RB1 ซึ่งอยู่บนแถบ q14 ของโครโมโซม 13 (13q14)
โดยปกติแต่ละเซลล์จะมียีน RB1 สองตัว:
- หากยีนทำงานอย่างถูกต้องอย่างน้อยหนึ่งสำเนา เรติโนบลาสโตมาจะไม่เกิดขึ้น (แต่ความเสี่ยงเพิ่มขึ้น)
- เมื่อยีนทั้งสองสำเนากลายพันธุ์หรือหายไป การเพิ่มจำนวนเซลล์ที่ไม่สามารถควบคุมได้จะเกิดขึ้น
ในหลายกรณี ยังไม่ชัดเจนว่าอะไรทำให้เกิดการเปลี่ยนแปลงในยีน RB1 (เรติโนบลาสโตมาประปราย); สิ่งเหล่านี้อาจเป็นผลมาจากข้อผิดพลาดทางพันธุกรรมแบบสุ่ม ซึ่งเกิดขึ้นได้ เช่น ในระหว่างการสืบพันธุ์และการแบ่งเซลล์ อย่างไรก็ตาม เป็นที่ทราบกันดีอยู่แล้วว่าความผิดปกติทางพันธุกรรมที่เป็นสาเหตุของเรติโนบลาสโตมานั้นยังสามารถส่งต่อจากพ่อแม่สู่ลูกได้ ด้วยรูปแบบการถ่ายทอดทางพันธุกรรมที่โดดเด่นแบบออโตโซม ซึ่งหมายความว่าหากผู้ปกครองมียีนกลายพันธุ์ (ยีนเด่น) เด็กแต่ละคนจะมีโอกาสได้รับมรดก 50% และมีโอกาส 50% ที่จะมีการสร้างยีนปกติ (ยีนด้อย)
- เซลล์เป็นครั้งคราวจะหยุดสำเนาปกติของยีน RB1 เท่านั้น (มีการกลายพันธุ์หนึ่งสำเนาแล้ว);
- การสูญเสีย RB1 สองสำเนาทำให้เกิด "การงอกของเรตินามากเกินไป
- เซลล์บางครั้งทำให้ยีน RB1 ปกติหยุดทำงาน
- สำเนาที่สองของยีน RB1 ถูกปิดใช้งาน
- การสูญเสีย RB1 สองสำเนาทำให้เกิดการเพิ่มจำนวนเซลล์มากเกินไปซึ่งนำไปสู่เรติโนบลาสโตมา
ลักษณะทางพันธุกรรมและโมเลกุล
- เรติโนบลาสโตมาเป็นเนื้องอกชนิดแรกที่เกี่ยวข้องโดยตรงกับ "ความผิดปกติทางพันธุกรรม (การลบหรือการกลายพันธุ์ของแถบ q14 ของโครโมโซม 13)
- RB1 เข้ารหัสโปรตีน pRb ซึ่งมีบทบาทสำคัญในวัฏจักรเซลล์: ช่วยให้การจำลองดีเอ็นเอและความก้าวหน้าของวัฏจักรเซลล์ มีส่วนร่วมในการควบคุมการถอดรหัสของยีน S phase (G1 → † "S)
- นอกจากเรติโนบลาสโตมาแล้ว ยีน RB1 ยังถูกปิดใช้งานในมะเร็งกระเพาะปัสสาวะ มะเร็งเต้านม และปอดอีกด้วย
เรติโนบลาสโตมาตามกรรมพันธุ์
เด็กที่เป็นโรคเรติโนบลาสโตมาที่ถ่ายทอดทางพันธุกรรมมักจะเป็นโรคนี้ตั้งแต่อายุยังน้อยมากกว่าผู้ป่วยประปราย นอกจากนี้ เด็กเหล่านี้มีความเสี่ยงที่จะเป็นมะเร็งที่ไม่เกี่ยวกับตาเพิ่มขึ้น เนื่องจากความผิดปกติในยีน RB1 นั้นมีมาแต่กำเนิด (กล่าวคือ มีตั้งแต่แรกเกิด) และส่งผลต่อเซลล์ทั้งหมดในร่างกาย (เรียกว่าการกลายพันธุ์ของเจิร์มไลน์) รวมทั้งเซลล์ทั้งสอง เรตินา: ด้วยเหตุนี้ เด็กที่มีรูปแบบการถ่ายทอดทางพันธุกรรมมักมีเรติโนบลาสโตมาทวิภาคีมากกว่าตาข้างเดียว
อาการ
เรียนรู้เพิ่มเติม: อาการจอประสาทตา
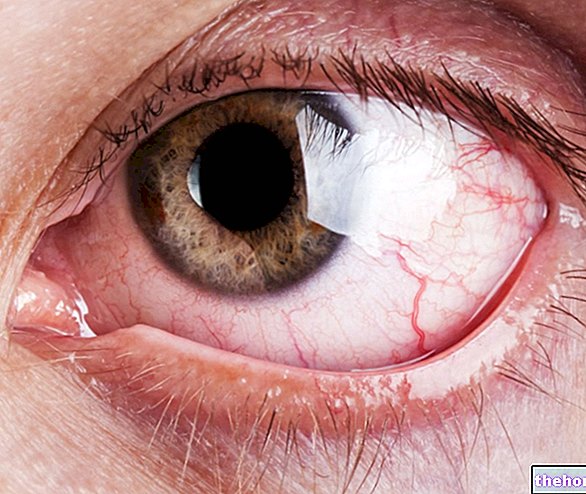
สัญญาณที่พบบ่อยและชัดเจนที่สุดของเรติโนบลาสโตมาคือลักษณะที่ผิดปกติของรูม่านตาซึ่งแสดงภาพสะท้อนสีเทาอมเทาเมื่อถูกลำแสงกระทบ (leukocoria หรือ amaurotic cat reflex). อาการและอาการแสดงอื่นๆ ได้แก่ การมองเห็นลดลง ปวดตาและตาแดง และพัฒนาการล่าช้า เด็กบางคนที่มีเรติโนบลาสโตมาอาจมีอาการเหล่ (ตาไม่ตรง) ในกรณีอื่น ๆ เป็นไปได้ที่จะพบโรคต้อหินในหลอดเลือดซึ่งหลังจากนั้นบางครั้งอาจทำให้ตาโต (buftalmo)
เซลล์มะเร็งสามารถบุกรุกดวงตาและโครงสร้างอื่นๆ ต่อไปได้:
- เรติโนบลาสโตมาในลูกตา เรติโนบลาสโตมาสามารถกำหนดเป็นลูกตาได้เมื่อเนื้องอกอยู่ภายในตาทั้งหมด เนื้องอกสามารถพบได้เฉพาะในเรตินาหรือส่งผลกระทบต่อส่วนอื่น ๆ เช่นคอรอยด์ ร่างกายปรับเลนส์ และส่วนหนึ่งของเส้นประสาทตา เรติโนบลาสโตมาในลูกตาจึงไม่แพร่กระจายไปยังเนื้อเยื่อรอบนอกดวงตา
- เรติโนบลาสโตมานอกตา เนื้องอกสามารถแพร่กระจายไปส่งผลต่อเนื้อเยื่อรอบดวงตา (เรติโนบลาสโตมาโคจร). มะเร็งยังสามารถแพร่กระจายไปยังส่วนอื่นๆ ของร่างกาย เช่น สมอง กระดูกสันหลัง ไขกระดูก และต่อมน้ำเหลือง (เรติโนบลาสโตมาระยะแพร่กระจาย).
การปรากฏตัวของการยืดออกของวงโคจร, การมีส่วนร่วมของ uveal และการบุกรุกของเส้นประสาทตาเป็นปัจจัยเสี่ยงที่ทราบกันดีอยู่แล้วสำหรับการพัฒนาของ metastatic retinoblastoma
การวินิจฉัย
ในกรณีที่มีประวัติครอบครัวเป็นบวก ผู้ป่วยจะได้รับการตรวจตาเพื่อตรวจคัดกรองมะเร็งเป็นประจำ หากเรติโนบลาสโตมาแต่กำเนิดเป็นแบบทวิภาคี มักได้รับการวินิจฉัยในปีแรกของชีวิต ในขณะที่เมื่อเกิดกับตาเพียงข้างเดียว การมีอยู่ของเนื้องอกจะได้รับการยืนยันเมื่ออายุประมาณ 18-30 เดือน
การวินิจฉัยทางคลินิกของเรติโนบลาสโตมาเกิดขึ้นโดยการตรวจอวัยวะ เนื้องอก ขึ้นอยู่กับตำแหน่ง อาจมองเห็นได้ในระหว่างการตรวจตาอย่างง่าย ๆ ผ่านการส่องกล้องตรวจทางอ้อม เทคนิคการถ่ายภาพสามารถใช้เพื่อยืนยันการวินิจฉัย กำหนดระยะของเนื้องอก (อยู่ที่ใด แพร่กระจายไปมากเพียงใด ไม่ว่าจะส่งผลต่อการทำงานของอวัยวะอื่นๆ ในร่างกาย เป็นต้น) และพิจารณาว่าการรักษาได้ผลหรือไม่ . การตรวจสอบอาจรวมถึงอัลตราซาวนด์ การตรวจเอกซเรย์คอมพิวเตอร์ (CT) และการถ่ายภาพด้วยคลื่นสนามแม่เหล็ก (MRI)
การวินิจฉัยระดับโมเลกุลและพันธุกรรมเป็นไปได้โดยการระบุการกลายพันธุ์ของยีน RB1 การวิเคราะห์ทางเซลล์ (เช่น โครโมโซม) ของลิมโฟไซต์ในเลือดส่วนปลายใช้เพื่อตรวจหาการลบหรือการจัดเรียงใหม่ที่เกี่ยวข้องกับโครโมโซม 13 (13q14.1-q14. 2) .
การรักษา
ในกรณีของเรติโนบลาสโตมา สามารถใช้วิธีการรักษาได้หลายอย่าง
วัตถุประสงค์ของการรักษาคือ:
- กำจัดเนื้องอกและช่วยชีวิตผู้ป่วย
- รักษาตาถ้าเป็นไปได้;
- รักษาวิสัยทัศน์ให้มากที่สุด
- หลีกเลี่ยงการพัฒนาของมะเร็งชนิดอื่นๆ ซึ่งอาจเกิดจากการรักษาได้เช่นกัน โดยเฉพาะในเด็กที่มีเรติโนบลาสโตมาที่ถ่ายทอดทางพันธุกรรม
การพยากรณ์โรค (ความน่าจะเป็นของการฟื้นตัว) และตัวเลือกการรักษาขึ้นอยู่กับปัจจัยต่อไปนี้:
- ระยะของเนื้องอก;
- อายุของผู้ป่วยและภาวะสุขภาพโดยทั่วไป
- ตำแหน่ง ขนาด และจำนวนของจุดโฟกัสของเนื้องอก
- การแพร่กระจายของมะเร็งไปยังบริเวณอื่นนอกจากลูกตา
- เป็นไปได้มากน้อยเพียงใดที่จะรักษาการมองเห็นไว้ในตาข้างเดียวหรือทั้งสองข้าง
กรณีส่วนใหญ่ของเรติโนบลาสโตมาได้รับการวินิจฉัยตั้งแต่เนิ่นๆ และรักษาได้สำเร็จ ก่อนที่มะเร็งจะลุกลามออกไปนอกลูกตา ส่งผลให้มีอัตราการหายขาดมากกว่า 90%
















.jpg)