ฟีนิลคีโตนูเรีย คืออะไร
ที่นั่น ฟีนิลคีโตนูเรีย (P.K.U.) มันคือโรคเมตาบอลิซึมที่ถ่ายทอดทางพันธุกรรมแบบถอยอัตโนมัติซึ่งส่งผลกระทบต่อ 1 ใน 10,000 คนและดูเหมือนว่าจะเกิดขึ้นใน homozygosity มากกว่าใน heterozygotes
ในกลุ่มของ hyperphenylalaninemia, phenylketonuria ส่งผลต่อการเผาผลาญของ phenylalanine อย่างมีนัยสำคัญและโดยเฉพาะอย่างยิ่ง แปลงเป็น ไทโรซีน; ฟีนิลคีโตนูเรียได้รับการยอมรับจากระดับฟีนิลอะลานีนในปัสสาวะและอนุพันธ์บางอย่าง
ภาวะแทรกซ้อนที่ร้ายแรงที่สุดของฟีนิลคีโตนูเรียคือ จิตล่าช้า.
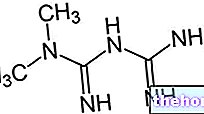
ฟีนิลอะลานีน ไทโรซีนและอนุพันธ์
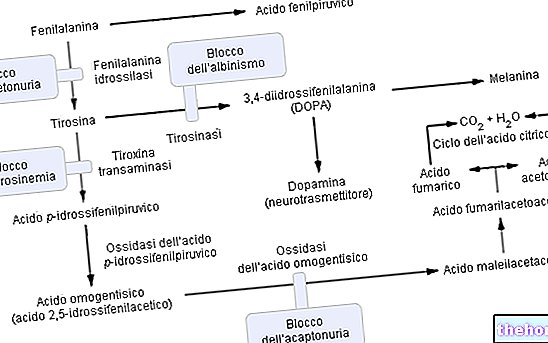
ฟีนิลอะลานีนเป็นกรดอะมิโนที่จำเป็นและเป็นโปรตีนในอาหารส่วนใหญ่ สามารถเปลี่ยนได้ด้วยเอ็นไซม์ ฟีนิลอะลานีนไฮดรอกซีเลส ในไทโรซีน (โดยการเพิ่มหมู่ไฮดรอกซิล -OH) ในทางกลับกัน ไทโรซีนเป็นสารตั้งต้นของกรดอะมิโนสำหรับการสังเคราะห์:
- L-DOPA (การสังเคราะห์โดปามีนระดับกลาง)
- อะดรีนาลีน
- Norepinephrine (สารสื่อประสาททั้งหมด)

กลไกการเกิดฟีนิลคีโตนูเรีย (P.K.U.)
ตามที่คาดไว้ ในฟีนิลคีโตนูเรีย เนื่องจากการกลายพันธุ์ของโครโมโซมหนึ่งหรือมากกว่า (6 ตัว) การแสดงออก (ด้วยเหตุนี้กิจกรรมการเผาผลาญ) ของฟีนิลอะลานีนไฮดรอกซิเลสจึงแทบไม่มีเลย การเปลี่ยนแปลงเหล่านี้สามารถมีได้หลายประเภท (จากการเปลี่ยนแปลงที่ "เข้าใจผิด" เป็นข้อบกพร่อง "การประกบ" หรือแม้แต่ "การลบบางส่วน") แต่สิ่งที่สำคัญคือเนื่องจากความไร้ประสิทธิภาพของเอนไซม์นี้ ระดับฟีนิลอะลานีนในเลือด (ซึ่งปกติคือ 1 มก. / 100 มล.) ในฟีนิลคีโตนูเรียที่โดดเด่น พวกเขาเข้าถึงปริมาณได้ง่ายยิ่งขึ้นถึง 50 เท่า
การทำงานของเอนไซม์ฟีนิลอะลานีนไฮดรอกซีเลส: ในการผลิตไทโรซีน (+ dihydrobiopterin), phenylalanine hydroxylase ต้องการ: phenylalanine, ออกซิเจนและ tetrahydrobiopterin (pteridine ลดลงซึ่งทำหน้าที่เป็น coofactor); ปฏิกิริยายังสามารถย้อนกลับได้และสามารถเปลี่ยนไดไฮโดรไบโอปเทอรินได้ใหม่ (ด้วยเอนไซม์ ไดไฮโดรปเทอริน รีดักเตส) ในเตตระไฮโดรไบโอปเทอริน
ภาวะแทรกซ้อน
Phenylketonuria สามารถก่อให้เกิดภาวะแทรกซ้อนรุนแรงมากหรือน้อยขึ้นอยู่กับความรุนแรงของอาการทางพยาธิวิทยาและความทันท่วงทีของการวินิจฉัย เป็นพยาธิสภาพทางพันธุกรรม phenylketonuria มีความโดดเด่นใน:
- เด่น ดังนั้นจึงโดดเด่นด้วยการไม่มีการใช้งานของเอนไซม์ฟีนิลอะลานีนไฮดรอกซีเลสอย่างสมบูรณ์
- Recessive ซึ่งมีเพียง 30% ของมรดกทางเอนไซม์ทั้งหมดที่ทำงานอยู่
ภาวะแทรกซ้อนของฟีนิลคีโตนูเรียนั้นมาจากสาเหตุและเป็นสัดส่วนโดยตรงกับการสะสมของฟีนิลอะลานีน อนุพันธ์ และการสังเคราะห์ที่ลดลงของไทโรซีน ในทางพยาธิวิทยา ฟีนิลอะลานีนส่วนเกินจะถูกกรองโดยไตที่ค่อนข้างมีประสิทธิภาพซึ่งดูดซับกลับเพียงบางส่วนเท่านั้น กำจัดด้วยปัสสาวะ อย่างไรก็ตาม การคงอยู่ของระดับของ hyper-phenylalaninemia เป็นตัวกำหนดปฏิกิริยาเมตาบอลิซึมของการเปลี่ยนแปลงระดับโมเลกุลใน กรดฟีนิลไพรูวิก และ/หรืออนุพันธ์อื่นๆ ระบายได้ง่ายขึ้น (phenylpyruvate, phenylacetate, phenylactate)
สิ่งที่ทำให้ฟีนิลคีโตนูเรียซับซ้อนคือความเป็นพิษของฟีนิลอะลานีน กรดฟีนิลไพรูวิก และอนุพันธ์ต่อระบบประสาทส่วนกลาง (CNS) การปรากฏตัวของมันมากเกินไปในการพัฒนาสมองอย่างไม่ลดละกำหนดรูปแบบของความบกพร่องทางสติปัญญา
หมายเหตุ ความเข้มข้นในพลาสมาของกรดอะมิโนอื่นๆ ลดลงเล็กน้อย อาจเป็นเพราะผลสะท้อนกลับเกี่ยวกับการดูดซึมในลำไส้หรือการดูดซึมซ้ำของท่อไต
ความเสียหายของสมองซึ่งเป็นภาวะแทรกซ้อนที่ร้ายแรงของฟีนิลคีโตนูเรียนั้นเกิดจากการลบกรดอะมิโนที่จำเป็นอื่น ๆ ในการสังเคราะห์โปรตีน Phenylketonuria - ไม่สามารถมองเห็นได้ทันทีหลังคลอด แต่หลังจากผ่านไปสองสามปี - หากไม่ได้รับการรักษา ต้องรักษาตัวในโรงพยาบาลของเด็กและไม่สามารถย้อนกลับได้อย่างสมบูรณ์
ฟีนิลคีโตนูเรียขั้นสูงสามารถมองเห็นได้ด้วยตาเปล่าอย่างชัดเจน ฟีนิลอะลานีนที่มีความเข้มข้นสูง ยับยั้งการทำงานของเอ็นไซม์ ไทโรซิเนส, บั่นทอนการสังเคราะห์เมลานินอย่างมีนัยสำคัญโดยการลดสีผิวและผม pigmentation; นอกจากนี้ การสะสมของ phenylacetate ในเส้นผมและผิวหนังทำให้ phenylketonurics มี "กลิ่นของหนู" ที่รุนแรงและไม่เป็นที่พอใจ














.jpg)



