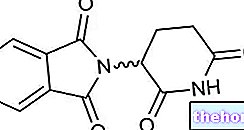
สารออกฤทธิ์: Abiraterone (Abiraterone acetate)
ไซทีก้า 250 มก. เม็ด
เหตุใดจึงใช้ Zytiga มีไว้เพื่ออะไร?
ไซทีก้ามียาที่เรียกว่าอะบิเรเทอโรนอะซิเตท ใช้รักษามะเร็งต่อมลูกหมากในผู้ใหญ่ที่แพร่กระจายไปยังส่วนอื่นๆ ของร่างกาย ไซทีก้าหยุดร่างกายไม่ให้สร้างฮอร์โมนเทสโทสเตอโรน ซึ่งสามารถชะลอการเติบโตของมะเร็งต่อมลูกหมากได้
เมื่อคุณทานยานี้ แพทย์ของคุณจะสั่งยาอีกตัวหนึ่งที่เรียกว่าเพรดนิโซนหรือเพรดนิโซโลน ยานี้ใช้เพื่อลดความเป็นไปได้ที่จะมีความดันโลหิตสูง น้ำในร่างกายมากเกินไป (การกักเก็บของเหลว) หรือมีระดับสารเคมีในเลือดต่ำที่เรียกว่าโพแทสเซียม
ข้อห้าม เมื่อไม่ควรใช้ Zytiga
ห้ามใช้ไซทีก้า
- หากคุณแพ้ abiraterone acetate หรือส่วนผสมอื่นใดของยานี้ (ระบุไว้ในหัวข้อ 6)
- ถ้าคุณเป็นผู้หญิง โดยเฉพาะถ้าคุณกำลังตั้งครรภ์ การใช้ไซทีก้ามีไว้สำหรับผู้ชายเท่านั้น
- หากคุณมีความเสียหายของตับอย่างรุนแรง
อย่าใช้ยานี้หากสิ่งเหล่านี้มีผลกับคุณ หากคุณไม่แน่ใจ ให้ปรึกษาแพทย์หรือเภสัชกรก่อนใช้ยานี้
ข้อควรระวังในการใช้งาน สิ่งที่คุณต้องรู้ก่อนใช้ยาไซติก้า
พูดคุยกับแพทย์หรือเภสัชกรของคุณก่อนรับประทานยานี้:
- หากคุณมีปัญหาเกี่ยวกับตับ
- หากคุณได้รับแจ้งว่าคุณมีความดันโลหิตสูงหรือหัวใจล้มเหลว หรือมีโพแทสเซียมในเลือดต่ำ (โพแทสเซียมในระดับต่ำอาจเพิ่มความเสี่ยงต่อปัญหาจังหวะการเต้นของหัวใจ)
- หากคุณเคยมีปัญหาอื่นๆ เกี่ยวกับหัวใจหรือหลอดเลือดของคุณ
- หากคุณมีหัวใจเต้นผิดปกติหรือเร็ว
- ถ้าคุณหายใจไม่ออก
- หากคุณน้ำหนักขึ้นอย่างรวดเร็ว
- หากคุณมีอาการบวมที่เท้า ข้อเท้า หรือขา
- หากคุณเคยทานยาที่เรียกว่า ketoconazole มาก่อนสำหรับมะเร็งต่อมลูกหมาก
- เกี่ยวกับความจำเป็นในการใช้ยานี้ร่วมกับเพรดนิโซนหรือเพรดนิโซโลน
- เกี่ยวกับผลกระทบที่อาจเกิดขึ้นกับกระดูก
- หากคุณมีระดับน้ำตาลในเลือดสูง
แจ้งแพทย์หากคุณได้รับแจ้งว่าคุณมีปัญหาเกี่ยวกับหัวใจหรือหลอดเลือด รวมถึงปัญหาจังหวะการเต้นของหัวใจ (ภาวะหัวใจเต้นผิดจังหวะ) หรือหากคุณกำลังรับการรักษาด้วยยาสำหรับอาการเหล่านี้
แจ้งให้แพทย์ประจำตัวของคุณทราบ หากคุณมีผิวหรือตาเหลือง ปัสสาวะสีเข้ม หรือคลื่นไส้หรืออาเจียนรุนแรง เนื่องจากอาจเป็นสัญญาณหรืออาการของปัญหาตับ ไม่ค่อยมีปัญหาเกี่ยวกับการทำงานของตับ (เรียกว่าภาวะตับวายเฉียบพลัน) ซึ่งอาจนำไปสู่ความตายได้
เซลล์เม็ดเลือดแดงลดลงความต้องการทางเพศลดลง (ความใคร่) กล้ามเนื้ออ่อนแรงและ / หรือปวดกล้ามเนื้ออาจเกิดขึ้น
หากคุณไม่แน่ใจว่าข้อใดข้อหนึ่งข้างต้นตรงกับคุณหรือไม่ ให้ปรึกษาแพทย์หรือเภสัชกรก่อนใช้ยานี้
การตรวจเลือด
ไซทีก้าสามารถส่งผลต่อตับและอาจไม่มีอาการ เมื่อคุณทานยานี้ แพทย์ของคุณจะตรวจเลือดเป็นระยะเพื่อตรวจหาผลของไซทีก้าต่อตับ
เด็กและวัยรุ่น
ยานี้ไม่ได้ระบุไว้ในเด็กและวัยรุ่น หากเด็กหรือวัยรุ่นกลืนกินไซทีก้าโดยไม่ได้ตั้งใจ ให้ไปโรงพยาบาลทันทีและนำแผ่นพับบรรจุภัณฑ์ไปด้วยเพื่อแสดงต่อแพทย์ประจำห้องฉุกเฉิน
ปฏิกิริยา ยาหรืออาหารชนิดใดที่สามารถเปลี่ยนผลของไซติกาได้
ปรึกษาแพทย์หรือเภสัชกรก่อนรับประทานยาใดๆ
แจ้งให้แพทย์หรือเภสัชกรทราบ หากคุณกำลังรับประทาน เพิ่งกำลังรับประทาน หรืออาจกำลังใช้ยาอื่นอยู่ นี่เป็นสิ่งสำคัญเพราะไซทีก้าสามารถเพิ่มผลของยาบางชนิดได้ เช่น ยารักษาโรคหัวใจ ยากล่อมประสาท ยาสมุนไพร (เช่น สาโทเซนต์จอห์น) และอื่นๆ แพทย์ของคุณอาจตัดสินใจเปลี่ยนขนาดยาเหล่านี้ นอกจากนี้ ยาบางชนิดยังสามารถเพิ่มหรือลดได้ ผลของไซทีก้าซึ่งอาจนำไปสู่ผลข้างเคียงหรือไซทีก้าอาจไม่ได้ผลดีเท่าที่ควร
ยาอื่นๆ ที่รับประทานร่วมกับไซทีก้า
การรักษาด้วยการกีดกันแอนโดรเจนสามารถเพิ่มความเสี่ยงต่อปัญหาจังหวะการเต้นของหัวใจ
แจ้งแพทย์หากคุณกำลังใช้ยาใดๆ:
- ใช้ในการรักษาปัญหาจังหวะการเต้นของหัวใจ (เช่น quinidine, procainamide, amiodarone และ sotalol);
- เป็นที่ทราบกันดีว่าเพิ่มความเสี่ยงต่อปัญหาจังหวะการเต้นของหัวใจ [เช่น เมธาโดน (ใช้บรรเทาอาการปวดและรักษาอาการติดยา), ม็อกซิฟลอกซาซิน (ยาปฏิชีวนะ), ยารักษาโรคจิต (ใช้สำหรับอาการป่วยทางจิตขั้นรุนแรง)]
ไซทีก้าพร้อมอาหาร
- ห้ามรับประทานยานี้ร่วมกับอาหาร (ดูหัวข้อ "การใช้ยานี้")
- การกินไซทีก้าร่วมกับอาหารอาจทำให้เกิดผลข้างเคียงได้
คำเตือน สิ่งสำคัญคือต้องรู้ว่า:
การตั้งครรภ์และให้นมบุตร
การใช้ไซทีก้าไม่ได้ระบุไว้ในผู้หญิง
- ยานี้อาจทำให้ทารกในครรภ์ได้รับอันตรายได้
- สตรีมีครรภ์หรือผู้ที่อาจจะตั้งครรภ์ควรสวมถุงมือหากจำเป็นต้องจับหรือจับไซทีก้า
- หากคุณมีเพศสัมพันธ์กับหญิงวัยเจริญพันธุ์ คุณควรใช้ถุงยางอนามัยและ "มาตรการคุมกำเนิดที่มีประสิทธิภาพ หากคุณมีเพศสัมพันธ์กับหญิงมีครรภ์ ให้ใช้ถุงยางอนามัยปกป้องทารกในครรภ์"
การขับรถและการใช้เครื่องจักร
ยานี้ไม่น่าจะส่งผลต่อความสามารถในการขับรถและใช้เครื่องมือหรือเครื่องจักรใดๆ ของคุณ
ไซทีก้าประกอบด้วยแลคโตสและโซเดียม
- ไซทีก้ามีแลคโตส (น้ำตาลชนิดหนึ่ง) หากคุณได้รับแจ้งจากแพทย์ว่าคุณแพ้น้ำตาลบางชนิด ให้ติดต่อแพทย์ก่อนใช้ยานี้
- ยานี้มีโซเดียมประมาณ 27 มก. ในขนาด 4 เม็ดต่อวัน สิ่งนี้ควรนำมาพิจารณาในผู้ป่วยที่รับประทานอาหารโซเดียมต่ำ
ปริมาณ วิธีการ และระยะเวลาในการบริหาร วิธีใช้ Zytiga: Posology
ใช้ยานี้ตามที่แพทย์ของคุณบอกเสมอ หากคุณไม่แน่ใจ ให้ปรึกษาแพทย์หรือเภสัชกรของคุณ
รับเท่าไหร่คะ
ปริมาณที่แนะนำคือ 1,000 มก. (สี่เม็ด) วันละครั้ง
กินยาตัวนี้
- กินยานี้ทางปาก
- ห้ามรับประทานไซทีก้าร่วมกับอาหาร
- รับประทานไซทีก้าหลังอาหารอย่างน้อยสองชั่วโมง และอย่ากินอะไรเป็นเวลาอย่างน้อยหนึ่งชั่วโมงหลังจากรับประทานไซทีก้า (ดูหัวข้อที่ 2" ไซทีก้าพร้อมอาหาร ")
- กลืนเม็ดทั้งหมดด้วยน้ำ
- อย่าทำลายแท็บเล็ต
- ไซทีก้าต้องรับประทานร่วมกับยาที่เรียกว่าเพรดนิโซนหรือเพรดนิโซโลน ใช้ยาเพรดนิโซนหรือเพรดนิโซโลนตามคำแนะนำของแพทย์
- คุณต้องทานเพรดนิโซนหรือเพรดนิโซโลนทุกวันขณะทานไซทีก้า
- อาจจำเป็นต้องเปลี่ยนปริมาณของ prednisone หรือ prednisolone ในกรณีฉุกเฉิน แพทย์ของคุณจะแนะนำคุณหากคุณต้องการเปลี่ยนปริมาณของ prednisone หรือ prednisolone ที่คุณใช้ อย่าหยุดรับประทานเพรดนิโซนหรือเพรดนิโซโลน เว้นแต่แพทย์จะสั่ง
แพทย์ของคุณอาจสั่งยาอื่นๆ ในขณะที่คุณรับประทานไซทีก้าและเพรดนิโซนหรือเพรดนิโซโลน
หากคุณลืมทานไซทีก้า
- หากคุณลืมกินไซทีก้าหรือเพรดนิโซนหรือเพรดนิโซโลน ให้ทานยาตามปกติในวันถัดไป
- หากคุณลืมรับประทานไซทีก้า หรือ เพรดนิโซน หรือ เพรดนิโซโลนเกินหนึ่งวัน ให้ปรึกษาแพทย์โดยไม่ต้องรอนานเกินไป
หากคุณหยุดทานไซทีก้า
อย่าหยุดรับประทานไซทีก้าหรือเพรดนิโซนหรือเพรดนิโซโลน เว้นแต่แพทย์จะแจ้งให้คุณทราบ
หากคุณมีคำถามเพิ่มเติมเกี่ยวกับการใช้ยานี้ ให้สอบถามแพทย์หรือเภสัชกรของคุณ
ยาเกินขนาด จะทำอย่างไรถ้าคุณทาน Zytiga มากเกินไป
หากคุณรับประทานไซทีก้ามากกว่าที่ควรจะเป็น ให้ปรึกษาแพทย์หรือไปโรงพยาบาลทันที
ผลข้างเคียง ผลข้างเคียงของ Zytiga . คืออะไร
เช่นเดียวกับยาอื่นๆ ยานี้อาจทำให้เกิดผลข้างเคียงได้ แม้ว่าจะไม่ใช่ทุกคนที่ได้รับก็ตาม
หากคุณสังเกตเห็นผลข้างเคียงใด ๆ ที่ระบุไว้ในเอกสารฉบับนี้ ให้หยุดใช้ไซทีก้าและติดต่อแพทย์ทันที:
- กล้ามเนื้ออ่อนแรง กล้ามเนื้อกระตุก หรือรู้สึกหัวใจเต้นแรง (ใจสั่น) สิ่งเหล่านี้อาจเป็นสัญญาณของโพแทสเซียมในเลือดต่ำ
ผลข้างเคียงอื่น ๆ ได้แก่ :
พบบ่อยมาก (อาจส่งผลกระทบมากกว่า 1 ใน 10 ผู้ป่วย)
ของเหลวในขาหรือเท้า ระดับโพแทสเซียมในเลือดต่ำ ความดันโลหิตสูง การติดเชื้อทางเดินปัสสาวะ ท้องร่วง
ร่วมกัน (อาจส่งผลกระทบถึง 1 ใน 10 ผู้ป่วย)
ระดับไขมันในเลือดสูง การทดสอบการทำงานของตับเพิ่มขึ้น เจ็บหน้าอก หัวใจเต้นผิดจังหวะ หัวใจล้มเหลว หัวใจเต้นเร็ว ติดเชื้อรุนแรงที่เรียกว่าภาวะติดเชื้อ กระดูกหัก อาหารไม่ย่อย เลือดในปัสสาวะ มีผื่น
ผิดปกติ (อาจส่งผลกระทบถึง 1 ใน 100 ผู้ป่วย)
ปัญหาเกี่ยวกับต่อมหมวกไต (เกี่ยวกับปัญหาเกลือและน้ำ) กล้ามเนื้ออ่อนแรง และ/หรือปวดกล้ามเนื้อ
หายาก (อาจส่งผลกระทบมากถึง 1 ใน 1,000 ผู้ป่วย)
การระคายเคืองต่อปอด (เรียกอีกอย่างว่าถุงลมอักเสบจากภูมิแพ้) ปัญหาเกี่ยวกับการทำงานของตับ (เรียกอีกอย่างว่าตับวายเฉียบพลัน)
ไม่ทราบ (ความถี่ไม่สามารถประมาณจากข้อมูลที่มีอยู่)
หัวใจวายการเปลี่ยนแปลงของ ECG - คลื่นไฟฟ้า (การยืด QT)
การสูญเสียกระดูกอาจเกิดขึ้นในผู้ชายที่รักษามะเร็งต่อมลูกหมาก ไซทีก้าร่วมกับเพรดนิโซนหรือเพรดนิโซโลนสามารถเพิ่มการสูญเสียมวลกระดูกได้
การรายงานผลข้างเคียง
หากคุณได้รับผลข้างเคียงใดๆ ให้ปรึกษาแพทย์หรือเภสัชกร ซึ่งรวมถึงผลข้างเคียงที่อาจเกิดขึ้นซึ่งไม่ได้ระบุไว้ในเอกสารฉบับนี้ คุณยังสามารถรายงานผลข้างเคียงได้โดยตรงผ่านระบบการรายงานระดับประเทศที่ระบุไว้ในภาคผนวก 5 โดยการรายงานผลข้างเคียง คุณสามารถช่วยให้ข้อมูลเพิ่มเติมเกี่ยวกับความปลอดภัยของยานี้ได้
การหมดอายุและการเก็บรักษา
- เก็บยานี้ให้พ้นสายตาและมือเด็ก
- ห้ามใช้ยานี้หลังจากวันหมดอายุซึ่งระบุไว้บนกล่องและฉลากขวด วันหมดอายุ หมายถึง วันสุดท้ายของเดือนนี้
- เก็บที่อุณหภูมิต่ำกว่า 30 องศาเซลเซียส
- ห้ามทิ้งยาลงในน้ำหรือของเสียในครัวเรือน ถามเภสัชกรว่าควรทิ้งยาที่ไม่ได้ใช้แล้วอย่างไร ซึ่งจะช่วยปกป้องสิ่งแวดล้อม
กำหนดเวลา "> ข้อมูลอื่นๆ
ไซทีก้าประกอบด้วยอะไรบ้าง
- สารออกฤทธิ์คืออะบิเรเทอโรนอะซิเตท แต่ละเม็ดประกอบด้วย abiraterone acetate 250 มก.
- ส่วนผสมอื่นๆ ได้แก่ microcrystalline cellulose, croscarmellose sodium, lactose monohydrate; แมกนีเซียมสเตียเรต โพวิโดน (K29 / K32) ปราศจากคอลลอยด์ซิลิกาและโซเดียมลอริลซัลเฟต (ดูหัวข้อที่ 2 "ไซทีก้ามีแลคโตสและโซเดียม")
คำอธิบายลักษณะและลักษณะของไซทีก้าในแพ็ค
- เม็ดยา ZYTIGA เป็นรูปวงรี สีขาวหรือสีขาวนวล สลัก "AA250" ที่ด้านหนึ่ง
- ยาเม็ดนี้บรรจุในขวดพลาสติกที่มีฝาปิดแบบป้องกันเด็กที่เป็นพลาสติก แต่ละขวดมี 120 เม็ด แต่ละกล่องมีหนึ่งขวด
เอกสารแพ็คเกจที่มา: AIFA (หน่วยงานยาอิตาลี) เนื้อหาที่เผยแพร่ในเดือนมกราคม 2016 ข้อมูลที่แสดงอาจไม่ทันสมัย
หากต้องการเข้าถึงเวอร์ชันล่าสุด ขอแนะนำให้เข้าถึงเว็บไซต์ AIFA (Italian Medicines Agency) ข้อจำกัดความรับผิดชอบและข้อมูลที่เป็นประโยชน์
01.0 ชื่อผลิตภัณฑ์ยา -
ไซทีก้า 250 มก. เม็ด
02.0 องค์ประกอบเชิงคุณภาพและเชิงปริมาณ -
แต่ละเม็ดประกอบด้วย อะบิเรเทอโรน อะซิเตท 250 มก.
สารเพิ่มปริมาณที่ทราบผลกระทบ
แต่ละเม็ดประกอบด้วยแลคโตส 189 มก. และโซเดียม 6.8 มก.
สำหรับรายการสารปรุงแต่งทั้งหมด ดูหัวข้อ 6.1
03.0 รูปแบบเภสัชกรรม -
ยาเม็ด
เม็ดยารูปวงรีสีขาวหรือสีขาว (ยาว 15.9 มม. x กว้าง 9.5 มม.) แกะลาย AA250 ด้านหนึ่ง
04.0 ข้อมูลทางคลินิก -
04.1 ข้อบ่งชี้การรักษา -
ไซทีก้าแสดงร่วมกับเพรดนิโซนหรือเพรดนิโซโลนสำหรับ:
• การรักษามะเร็งต่อมลูกหมากที่ดื้อต่อระยะลุกลามในผู้ชายผู้ใหญ่ที่ไม่มีอาการหรือแสดงอาการไม่รุนแรงหลังจากความล้มเหลวของการบำบัดด้วยการกีดกันแอนโดรเจนและผู้ที่ยังไม่ได้ระบุการรักษาด้วยเคมีบำบัด (ดูหัวข้อ 5.1)
• การรักษามะเร็งต่อมลูกหมากที่ดื้อต่อการแพร่กระจายในระยะลุกลามในผู้ชายที่เป็นผู้ใหญ่ที่มีโรคลุกลามในระหว่างหรือหลังการรักษาด้วยเคมีบำบัดที่ใช้ docetaxel
04.2 วิทยาและวิธีการบริหาร -
ยานี้ควรกำหนดโดยแพทย์ผู้มีประสบการณ์ในการใช้ยาต้านมะเร็ง
ปริมาณ
ปริมาณที่แนะนำคือ 1,000 มก. (สี่เม็ด 250 มก.) ที่ต้องรับประทานในขณะท้องว่างเป็นปริมาณรายวันเพียงครั้งเดียว (ดู "วิธีการให้ยา" ด้านล่าง) การรับประทานยาเม็ดพร้อมอาหารส่งผลให้มีการได้รับ abiraterone อย่างเป็นระบบมากขึ้น (ดูหัวข้อ 4.5 และ 5.2)
ไซทีก้าควรรับประทานร่วมกับเพรดนิโซนหรือเพรดนิโซโลนในขนาดต่ำ ปริมาณที่แนะนำของ prednisone หรือ prednisolone คือ 10 มก. ต่อวัน
การตัดอัณฑะทางการแพทย์ด้วยแอนะล็อกของปัจจัยการปลดปล่อย gonadotropin (ลูทีไนซิ่งฮอร์โมนการปลดปล่อยฮอร์โมน, LHRH) ควรดำเนินต่อไปในระหว่างการรักษาในผู้ป่วยที่ไม่ได้ผ่าตัดตอน
ก่อนเริ่มการรักษา ควรวัดระดับ transaminase ในซีรัมทุกสองสัปดาห์ในช่วงสามเดือนแรกของการรักษาและทุกเดือนหลังจากนั้น ตรวจสอบความดันโลหิต โพแทสเซียมในเลือด และการกักเก็บของเหลวทุกเดือน (ดูหัวข้อ 4.4) อย่างไรก็ตาม ผู้ป่วยที่มีความเสี่ยงอย่างมีนัยสำคัญต่อภาวะหัวใจล้มเหลวควรได้รับการตรวจสอบทุก 2 สัปดาห์ในช่วงสามเดือนแรกของการรักษาและทุกเดือนหลังจากนั้น (ดูหัวข้อ 4.4)
พิจารณาการรักษาระดับโพแทสเซียม≥ 4.0 มิลลิโมลาร์ในผู้ป่วยที่มีภาวะโพแทสเซียมในเลือดต่ำหรือในผู้ที่มีภาวะโพแทสเซียมในเลือดต่ำในระหว่างการรักษาด้วยไซทีก้า
สำหรับผู้ป่วยที่มีความเป็นพิษระดับ ≥ 3 รวมถึงความดันโลหิตสูง ภาวะโพแทสเซียมในเลือดต่ำ อาการบวมน้ำ และความเป็นพิษอื่นๆ ที่ไม่ใช่แร่ไมเนราโลคอร์ติคอยด์ ควรหยุดการรักษาและให้การรักษาที่เหมาะสม ไม่ควรให้การรักษาด้วยไซทีก้าต่อจนกว่าอาการพิษจะลดลงเป็นระดับ 1 หรือการตรวจวัดพื้นฐาน
หากลืมรับประทานไซทีก้า, เพรดนิโซน หรือเพรดนิโซโลนในขนาดรายวัน การรักษาควรกลับมาดำเนินการใหม่ในวันถัดไป โดยใช้ขนาดยาตามปกติในแต่ละวัน
พิษต่อตับ
ในผู้ป่วยที่มีความเป็นพิษต่อตับระหว่างการรักษา (เพิ่ม alanine aminotransferase [ALT] หรือ aspartate aminotransferase [AST] มากกว่า 5 เท่าของค่าปกติ [ULN]) ควรหยุดการรักษาทันที (ดูหัวข้อ 4.4) การเริ่มต้นใหม่ของการรักษา หลังจากที่ค่าการทดสอบการทำงานของตับของผู้ป่วยกลับสู่การตรวจวัดพื้นฐานแล้ว สามารถทำได้โดยลดขนาดยาลง 500 มก. (สองเม็ด) วันละครั้ง ในผู้ป่วยที่ได้รับการบำบัดซ้ำ ควรตรวจสอบระดับ transaminase ในซีรัมอย่างน้อยทุกสองสัปดาห์เป็นเวลาสามเดือนและหลังจากนั้นทุกเดือน หากมีความเป็นพิษต่อตับเกิดขึ้นอีกเมื่อลดขนาดยาลง 500 มก. ต่อวัน, ต้องหยุดการรักษา
หากผู้ป่วยมีความเป็นพิษต่อตับอย่างรุนแรงในช่วงเวลาใดๆ ระหว่างการรักษา (ALT หรือ AST 20 เท่าของ ULN) ควรหยุดการรักษาและไม่ควรให้ผู้ป่วยได้รับการรักษาซ้ำ
การด้อยค่าของตับ
ในผู้ป่วยที่มีภาวะตับบกพร่องระดับอ่อนอยู่ก่อนแล้ว Child-Pugh Class A ไม่จำเป็นต้องปรับขนาดยา
การด้อยค่าของตับในระดับปานกลาง (Child-Pugh Class B) ส่งผลให้มีการได้รับ abiraterone อย่างเป็นระบบเพิ่มขึ้นประมาณสี่เท่าหลังจากได้รับ abiraterone acetate 1,000 มก. ในช่องปากครั้งเดียว (ดูหัวข้อ 5.2 ) ไม่มีข้อมูลทางคลินิกและความปลอดภัย ของ abiraterone acetate เมื่อให้แก่ผู้ป่วยที่มีความบกพร่องของตับในระดับปานกลางหรือรุนแรง (Child-Plugh Class B หรือ C) คาดว่าจะไม่มีการปรับขนาดยา การใช้ ZYTIGA ควรพิจารณาด้วยความระมัดระวังในผู้ป่วยที่มีความบกพร่องของตับในระดับปานกลางซึ่งผลประโยชน์จะต้องมีมากกว่าความเสี่ยงที่เป็นไปได้อย่างชัดเจน (ดูหัวข้อ 4.2 และ 5.2) ไม่ควรใช้ ZYTIGA ในผู้ป่วยที่มีความบกพร่องทางตับอย่างรุนแรง (ดูหัวข้อ 4.3, 4.4 และ 5.2)
การด้อยค่าของไต
ไม่จำเป็นต้องปรับขนาดยาในผู้ป่วยไตวาย (ดูหัวข้อ 5.2) อย่างไรก็ตาม ไม่มีประสบการณ์ทางคลินิกในผู้ป่วยมะเร็งต่อมลูกหมากและการด้อยค่าของไตอย่างรุนแรง ควรใช้ความระมัดระวังในผู้ป่วยเหล่านี้ (ดูหัวข้อ 4.4)
ประชากรเด็ก
ไม่มีข้อบ่งชี้สำหรับการใช้ไซทีก้าอย่างเฉพาะเจาะจงในประชากรเด็ก
วิธีการบริหาร
ไซทีก้าใช้สำหรับช่องปาก
ควรรับประทานยาเม็ดหลังอาหารอย่างน้อย 2 ชั่วโมง และห้ามรับประทานอาหารเป็นเวลาอย่างน้อย 1 ชั่วโมงหลังรับประทานยาเม็ด ควรกลืนกินทั้งเม็ดด้วยน้ำบางส่วน
04.3 ข้อห้าม -
- ภูมิไวเกินต่อสารออกฤทธิ์หรือสารเพิ่มปริมาณใด ๆ ที่ระบุไว้ในหัวข้อ 6.1
- สตรีมีครรภ์หรือมีบุตร (ดูหัวข้อ 4.6)
• การด้อยค่าของตับอย่างรุนแรง [Child-Plugh class C scale (ดูหัวข้อ 4.2, 4.4 และ 5.2)]
04.4 คำเตือนพิเศษและข้อควรระวังในการใช้งาน -
ความดันโลหิตสูง ภาวะโพแทสเซียมในเลือดต่ำ การกักเก็บของเหลว และภาวะหัวใจล้มเหลวที่เกิดจากแร่ธาตุคอร์ติคอยด์มากเกินไป
ไซทีก้าสามารถทำให้เกิดความดันโลหิตสูง ภาวะโพแทสเซียมในเลือดต่ำ และการกักเก็บของเหลว (ดูหัวข้อ 4.8) อันเป็นผลมาจากระดับแร่คอร์ติคอยด์ที่เพิ่มขึ้นซึ่งเกิดจากการยับยั้ง CYP17 (ดูหัวข้อ 5.1) การใช้ยาคอร์ติโคสเตียรอยด์ร่วมกันจะยับยั้งการทำงานของฮอร์โมนอะดรีโนคอร์ติโคทรอปิก (ACTH) ส่งผลให้อุบัติการณ์และความรุนแรงของอาการข้างเคียงเหล่านี้ลดลง ควรใช้ความระมัดระวังในการรักษาผู้ป่วยที่มีภาวะทางคลินิกที่อาจได้รับผลกระทบจากความดันโลหิตที่เพิ่มขึ้น จากภาวะโพแทสเซียมในเลือดต่ำ (เช่น ผู้ที่รักษาด้วย glycosides หัวใจ) หรือการกักเก็บของเหลว (เช่น ผู้ที่มีภาวะหัวใจล้มเหลว) ที่มีภาวะหลอดเลือดหัวใจตีบรุนแรงหรือไม่เสถียร กล้ามเนื้อหัวใจตายเฉียบพลันหรือภาวะหัวใจเต้นผิดจังหวะ และผู้ที่มีภาวะไตวายอย่างเข้มงวด
ไซทีก้าควรใช้ด้วยความระมัดระวังในผู้ป่วยที่เป็นโรคหัวใจและหลอดเลือด การทดลองทางคลินิกระยะที่ 3 ไม่รวมผู้ป่วยที่มีภาวะความดันโลหิตสูงที่ไม่สามารถควบคุมได้ โรคหัวใจที่มีนัยสำคัญทางคลินิกที่มีหลักฐานจากกล้ามเนื้อหัวใจตาย หรือเหตุการณ์ atherothrombotic ภายใน 6 เดือนที่ผ่านมา โรคหลอดเลือดหัวใจตีบรุนแรงหรือไม่เสถียร หรือภาวะหัวใจล้มเหลวระดับ III หรือ IV ของ สมาคมโรคหัวใจนิวยอร์ก (NYHA) (การศึกษา 301) หรือภาวะหัวใจล้มเหลวระดับ II - IV (การศึกษา 302) หรือการวัดภาวะหัวใจห้องบนเต้นผิดจังหวะแบบเศษส่วนดีดออกของหัวใจหรือภาวะหัวใจเต้นผิดจังหวะอื่น ๆ ที่ต้องได้รับการรักษาพยาบาล ความปลอดภัยในผู้ป่วยที่มีหัวใจห้องล่างซ้าย (LVEF)
ก่อนการรักษาผู้ป่วยที่มีความเสี่ยงสูงต่อภาวะหัวใจล้มเหลว (เช่น ประวัติของภาวะหัวใจล้มเหลว ความดันโลหิตสูงที่ไม่สามารถควบคุมได้ หรือเหตุการณ์เกี่ยวกับหัวใจ เช่น โรคหัวใจขาดเลือด) ควรพิจารณาการประเมินการทำงานของหัวใจ (เช่น echocardiogram) ก่อนการรักษาด้วย ZYTIGA ควรได้รับการรักษาเกี่ยวกับหัวใจ ความล้มเหลวและการทำงานของหัวใจที่เหมาะสม ความดันโลหิตสูง ภาวะโพแทสเซียมในเลือดต่ำ และการเก็บของเหลวควรได้รับการแก้ไขและควบคุม ในระหว่างการรักษา ความดันโลหิต ระดับโพแทสเซียมในเลือดและการกักเก็บของเหลว (การเพิ่มของน้ำหนัก อาการบวมน้ำที่บริเวณรอบข้าง) และอาการและอาการแสดงอื่นๆ ของภาวะหัวใจล้มเหลว 2 สัปดาห์ เป็นเวลา 3 เดือน แล้วเดือนละครั้ง และแก้ไขความผิดปกติ มีการสังเกตการยืดช่วง QT ในผู้ป่วยที่มีภาวะโพแทสเซียมในเลือดต่ำร่วมกับการรักษาด้วยยา ZYTIGA ประเมินการทำงานของหัวใจตามที่ระบุไว้ทางคลินิก กำหนดการจัดการที่เหมาะสม และพิจารณาหยุดการรักษานี้ในกรณีที่การทำงานของหัวใจลดลงอย่างมีนัยสำคัญ (ดูหัวข้อ 4.2)
ความเป็นพิษต่อตับและการด้อยค่าของตับ
ระดับเอนไซม์ในตับสูงขึ้นอย่างเห็นได้ชัดซึ่งนำไปสู่การหยุดชะงักของการรักษาหรือการปรับเปลี่ยนขนาดยาได้รับการสังเกตในการทดลองทางคลินิกที่มีการควบคุม (ดูหัวข้อ 4.8) ควรวัดระดับทรานสอะมิเนสในซีรัมทุกสองสัปดาห์ก่อนเริ่มการรักษา ในช่วง 3 เดือนแรกของการรักษาและทุกเดือน หลังจากนั้น หากมีอาการและอาการแสดงทางคลินิกที่บ่งชี้ถึงความเป็นพิษต่อตับ ควรตรวจวัดระดับ transaminases ในซีรัมทันที หาก ALT หรือ AST เพิ่มขึ้น 5 เท่า "ULN ควรหยุดการรักษาทันทีและควรติดตามการทำงานของตับอย่างใกล้ชิด . สามารถกลับมารักษาต่อได้ในปริมาณที่ลดลงหลังจากที่ค่าการทดสอบการทำงานของตับของผู้ป่วยกลับสู่การตรวจวัดพื้นฐานแล้ว (ดูหัวข้อ 4.2)
หากผู้ป่วยมีความเป็นพิษต่อตับอย่างรุนแรง (ALT หรือ AST เพิ่มขึ้น 20 เท่าของ ULN) ในระหว่างการรักษา ควรหยุดการรักษาและไม่ควรให้ผู้ป่วยดังกล่าวซ้ำ
ผู้ป่วยที่เป็นโรคตับอักเสบจากไวรัสที่ใช้งานหรือมีอาการไม่รวมอยู่ในการศึกษาทางคลินิก ดังนั้นจึงไม่มีข้อมูลสนับสนุนการใช้ไซทีก้าในประชากรกลุ่มนี้
ไม่มีข้อมูลเกี่ยวกับความปลอดภัยทางคลินิกและประสิทธิภาพของการให้ abiraterone acetate หลายขนาดเมื่อให้กับผู้ป่วยที่มีความผิดปกติของตับในระดับปานกลางหรือรุนแรง (Child-Plugh Class B หรือ C) การใช้ ZYTIGA ควรได้รับการประเมินด้วยความระมัดระวังในผู้ป่วย การด้อยค่าของตับในระดับปานกลางซึ่งผลประโยชน์ต้องมีมากกว่าความเสี่ยงที่เป็นไปได้อย่างชัดเจน (ดูหัวข้อ 4.2 และ 5.2) ห้ามใช้ไซทีก้าในผู้ป่วยที่มีความบกพร่องทางตับอย่างรุนแรง (ดูหัวข้อ 4.2, 4.3 และ 5.2)
มีรายงานหลังการขายที่ไม่ค่อยพบนักเกี่ยวกับภาวะตับวายเฉียบพลันและตับอักเสบขั้นรุนแรง ซึ่งบางรายอาจถึงแก่ชีวิตได้ (ดูหัวข้อ 4.8)
การยุติการให้ยาคอร์ติโคสเตียรอยด์และการรักษาสถานการณ์ตึงเครียด
แนะนำให้ใช้ความระมัดระวังและติดตามภาวะต่อมหมวกไตไม่เพียงพอหากผู้ป่วยหยุดการรักษาด้วย prednisone หรือ prednisolone หาก ZYTIGA ยังคงดำเนินต่อไปหลังจากหยุดยา corticosteroids ผู้ป่วยควรได้รับการตรวจสอบอาการของ
ในผู้ป่วยที่ได้รับ prednisone หรือ prednisolone ที่มีความเครียดผิดปกติ อาจแนะนำให้เพิ่มขนาดยา corticosteroids ก่อน ระหว่าง และหลังสถานการณ์ตึงเครียด
ความหนาแน่นของกระดูก
ความหนาแน่นของกระดูกลดลงอาจเกิดขึ้นในผู้ชายที่เป็นมะเร็งต่อมลูกหมากระยะลุกลาม (มะเร็งต่อมลูกหมากที่ดื้อต่อการตัดอัณฑะ) การใช้ไซทีก้าร่วมกับกลูโคคอร์ติคอยด์อาจเพิ่มประสิทธิภาพนี้ได้
การใช้คีโตโคนาโซลก่อนหน้านี้
ผู้ป่วยมะเร็งต่อมลูกหมากที่เคยรักษาด้วย ketoconazole อาจได้รับอัตราการตอบสนองที่ต่ำกว่า
น้ำตาลในเลือดสูง
การใช้กลูโคคอร์ติคอยด์สามารถเพิ่มระดับน้ำตาลในเลือดสูง ดังนั้นควรตรวจวัดระดับน้ำตาลในเลือดบ่อยครั้งในผู้ป่วยเบาหวาน
ใช้ในเคมีบำบัด
ความปลอดภัยและประสิทธิภาพของไซทีก้าที่ใช้ควบคู่กับเคมีบำบัดที่เป็นพิษต่อเซลล์ยังไม่เป็นที่ยอมรับ (ดูหัวข้อ 5.1)
การไม่ทนต่อสารเพิ่มปริมาณ
ยานี้มีแลคโตส ผู้ป่วยที่มีปัญหาทางพันธุกรรมที่หายากของการแพ้กาแลคโตส การขาด Lapp lactase หรือการดูดซึมน้ำตาลกลูโคส - กาแลคโตส malabsorption ไม่ควรรับประทานยานี้ นอกจากนี้ ยานี้มีโซเดียมมากกว่า 1 มิลลิโมล (หรือ 27.2 มก.) ในขนาดยาสี่เม็ด สิ่งนี้ควรพิจารณาสำหรับผู้ป่วยที่รับประทานอาหารโซเดียมต่ำ
ความเสี่ยงที่อาจเกิดขึ้น
ภาวะโลหิตจางและความผิดปกติทางเพศอาจเกิดขึ้นในผู้ชายที่เป็นมะเร็งต่อมลูกหมากที่ทนต่อระยะลุกลามได้ รวมถึงผู้ที่รับการรักษาด้วยไซทีก้า
ผลต่อกล้ามเนื้อโครงร่าง
มีรายงานกรณีของโรคกล้ามเนื้อหัวใจขาดเลือดในผู้ป่วยที่ได้รับการรักษาด้วยไซทีก้า ผู้ป่วยบางรายมีภาวะ rhabdomyolysis กับภาวะไตวาย กรณีส่วนใหญ่เกิดขึ้นภายในเดือนแรกของการรักษา และแก้ไขได้หลังจากหยุดไซทีก้า ผู้ป่วยควรระมัดระวังในการรักษาร่วมกับผลิตภัณฑ์ยาที่ทราบว่ามีความเกี่ยวข้องกับโรคกล้ามเนื้อหรือกล้ามเนื้อหัวใจขาดเลือด (rhabdomyolysis)
ปฏิกิริยากับยาอื่น ๆ
ควรหลีกเลี่ยงตัวกระตุ้นที่มีศักยภาพของ CYP3A4 ในระหว่างการรักษา เว้นแต่ไม่มีทางเลือกในการรักษา เนื่องจากความเสี่ยงของการได้รับ abiraterone ลดลง (ดูหัวข้อ 4.5)
04.5 ปฏิกิริยากับผลิตภัณฑ์ยาอื่น ๆ และรูปแบบอื่น ๆ ของการโต้ตอบ -
ผลของอาหารต่ออะบิเรเทอโรน อะซิเตท
การบริหารอาหารจะเพิ่มการดูดซึมของ abiraterone acetate อย่างมีนัยสำคัญ ยังไม่มีการกำหนดประสิทธิภาพและความปลอดภัยเมื่อให้พร้อมกับอาหาร ดังนั้น ไม่ควรรับประทานยานี้พร้อมกับอาหาร (ดูหัวข้อ 4.2 และ 5.2)
ปฏิสัมพันธ์กับผลิตภัณฑ์ยาอื่น ๆ
ศักยภาพสำหรับผลิตภัณฑ์ยาอื่น ๆ ที่มีผลต่อการได้รับ abiraterone
ในการศึกษาปฏิสัมพันธ์ทางเภสัชจลนศาสตร์ทางคลินิกในคนที่มีสุขภาพดีที่ได้รับยากระตุ้น CYP3A4 ที่มีศักยภาพ rifampicin 600 มก. ต่อวันเป็นเวลา 6 วัน ตามด้วย abiraterone acetate 1,000 มก. ครั้งเดียว ค่าเฉลี่ย AUC ในพลาสมาของ abiraterone ลดลง 55. %
สารกระตุ้นที่มีศักยภาพของ CYP3A4 (เช่น phenytoin, carbamazepine, rifampicin, rifabutin, rifapentine, phenobarbital, St. John's wort [Hypericum perforatum]) ควรหลีกเลี่ยงในระหว่างการรักษา เว้นแต่ไม่มีทางเลือกในการรักษา
ในการศึกษาปฏิสัมพันธ์ทางเภสัชจลนศาสตร์ทางคลินิกอื่นในอาสาสมัครที่มีสุขภาพดี การใช้ยา ketoconazole ซึ่งเป็นตัวยับยั้ง CYP3A4 ที่มีศักยภาพ ไม่มีผลอย่างมีนัยสำคัญทางคลินิกต่อเภสัชจลนศาสตร์ของ abiraterone
ศักยภาพที่จะมีอิทธิพลต่อการสัมผัสผลิตภัณฑ์ยาอื่น ๆ
Abiraterone เป็นตัวยับยั้งเอนไซม์ตับ CYP2D6 และ CYP2C8
ในการศึกษาเพื่อตรวจสอบผลของ abiraterone acetate (บวก prednisone) กับ dextromethorphan สารตั้งต้น CYP2D6 เพียงครั้งเดียวการได้รับ dextromethorphan อย่างเป็นระบบ (AUC) ของ dextromethorphan เพิ่มขึ้นประมาณ 2.9 เท่า AUC24 ต่อ dextrorphan ซึ่งเป็นสารออกฤทธิ์ของ dextromethorphan เพิ่มขึ้นประมาณ 33%
ควรใช้ความระมัดระวังในการบริหารผลิตภัณฑ์ยาที่กระตุ้นหรือเผาผลาญโดย CYP2D6 โดยเฉพาะผลิตภัณฑ์ยาที่มีดัชนีการรักษาต่ำ ควรพิจารณาการลดขนานยาของผลิตภัณฑ์ยาที่มีดัชนีการรักษาต่ำที่เผาผลาญโดย CYP2D6 ตัวอย่างของผลิตภัณฑ์ยาที่เผาผลาญโดย CYP2D6 ได้แก่ metoprolol, propranolol, desipramine, venlafaxine, haloperidol, risperidone, propafenone, flecanide, codeine, oxycodone และ tramadol (ยาสามตัวหลังต้องการกิจกรรม CYP2D6 สำหรับการก่อตัวของสารระงับปวดที่ใช้งานอยู่)
ในการศึกษาปฏิสัมพันธ์ระหว่างยาและยาทางคลินิกของ CYP2C8 ในคนที่มีสุขภาพดี AUC ของ pioglitazone เพิ่มขึ้น 46% และ AUC สำหรับ M-III และ M-IV ซึ่งเป็นสารออกฤทธิ์ของ pioglitazone ลดลง 10% เมื่อให้ pioglitazone ร่วมกับยาอะบิราเทอโรน อะซิเตทขนาด 1,000 มก. แม้ว่าผลลัพธ์เหล่านี้บ่งชี้ว่าไม่คาดว่าจะได้รับสารเพิ่มขึ้นอย่างมีนัยสำคัญทางคลินิกเมื่อใช้ร่วมกับยารักษาโรคที่ CYP2C8 กำจัดไป แต่ผู้ป่วยควรได้รับการตรวจสอบอย่างรอบคอบเพื่อหาสัญญาณของความเป็นพิษที่เกี่ยวข้องกับ CYP2C8 สารตั้งต้นที่มีดัชนีการรักษาที่แคบเมื่อใช้ควบคู่กัน
ในหลอดทดลองพบว่าสารเมแทบอไลต์ที่สำคัญของ abiraterone sulfate และ N-oxide abiraterone sulfate สามารถยับยั้งการลำเลียงของการดูดซึม OATP1B1 ของตับและด้วยเหตุนี้อาจทำให้ความเข้มข้นของผลิตภัณฑ์ยาที่กำจัดโดย OATP1B1 เพิ่มขึ้น ไม่มีข้อมูลทางคลินิกเพื่อยืนยันการมีปฏิสัมพันธ์กับผู้ขนส่ง
ใช้กับผลิตภัณฑ์ยาที่ขึ้นชื่อในการยืดช่วง QT
เนื่องจากการบำบัดด้วยการกีดกันแอนโดรเจนสามารถยืดช่วง QT ได้ ควรใช้ความระมัดระวังในการบริหารไซทีก้าร่วมกับผลิตภัณฑ์ยาที่ทราบว่ายืดช่วง QT หรือผลิตภัณฑ์ทางยาที่สามารถกระตุ้นให้เกิด torsade de pointes เช่น ยาลดความอ้วนแบบ IA (เช่น quinidine, disopyramide) หรือคลาส III (เช่น amiodarone, sotalol, dofetilide, ibutilide), methadone, moxifloxacin, antipsychotics เป็นต้น
ใช้ร่วมกับสไปโรโนแลคโตน
Spironolactone จับตัวรับแอนโดรเจนและสามารถเพิ่มระดับแอนติเจนจำเพาะต่อมลูกหมาก (PSA) ได้ ไม่แนะนำให้ใช้ร่วมกับไซทีก้า (ดูหัวข้อ 5.1)
04.6 การตั้งครรภ์และให้นมบุตร -
ผู้หญิงที่มีศักยภาพในการคลอดบุตร
ไม่มีข้อมูลเกี่ยวกับการใช้ไซทีก้าในสตรีมีครรภ์ และการใช้ยานี้มีข้อห้ามในสตรีมีครรภ์
การคุมกำเนิดในผู้ชายและผู้หญิง
ไม่ทราบว่า abiraterone หรือสารเมตาบอลิซึมถูกขับออกมาในน้ำอสุจิหรือไม่ หากผู้ป่วยมีเพศสัมพันธ์กับผู้หญิงในระหว่างตั้งครรภ์ แนะนำให้ใช้ถุงยางอนามัย หากผู้ป่วยมีเพศสัมพันธ์กับสตรีมีครรภ์ ขอแนะนำให้ใช้ถุงยางอนามัยร่วมกับมาตรการคุมกำเนิดอื่นที่มีประสิทธิผล การศึกษาในสัตว์ทดลองแสดงความเป็นพิษต่อการเจริญพันธุ์ (ดูหัวข้อ 5.3)
การตั้งครรภ์
ไซทีก้าไม่ได้ระบุไว้ในสตรีและมีข้อห้ามในระหว่างตั้งครรภ์หรือในสตรีมีครรภ์ (ดูหัวข้อ 4.3 และ 5.3)
เวลาให้อาหาร
ห้ามใช้ไซทีก้าในสตรี
ภาวะเจริญพันธุ์
Abiraterone มีผลต่อภาวะเจริญพันธุ์ในหนูเพศผู้และเพศเมีย แต่ผลกระทบเหล่านี้สามารถย้อนกลับได้อย่างสมบูรณ์ (ดูหัวข้อ 5.3)
04.7 ผลกระทบต่อความสามารถในการขับขี่และการใช้เครื่องจักร -
ไซทีก้าไม่มีหรือมีอิทธิพลเล็กน้อยต่อความสามารถในการขับขี่และการใช้เครื่องจักร
04.8 ผลกระทบที่ไม่พึงประสงค์ -
สรุปข้อมูลความปลอดภัย
อาการไม่พึงประสงค์ที่สังเกตได้บ่อยที่สุดคืออาการบวมน้ำที่บริเวณรอบข้าง ภาวะโพแทสเซียมในเลือดต่ำ ความดันโลหิตสูง และการติดเชื้อทางเดินปัสสาวะ
อาการไม่พึงประสงค์ที่สำคัญอื่นๆ ได้แก่ โรคหัวใจ ความเป็นพิษต่อตับ กระดูกหัก และถุงลมอักเสบจากภูมิแพ้
ไซทีก้าสามารถทำให้เกิดความดันโลหิตสูง ภาวะโพแทสเซียมในเลือดต่ำ และการกักเก็บของเหลวอันเป็นผลมาจากกลไกการออกฤทธิ์ทางเภสัชพลศาสตร์ ในการศึกษาทางคลินิก อาการไม่พึงประสงค์ที่คาดว่าจะได้รับจากไมเนอรัลคอร์ติคอยด์มักพบในผู้ป่วยที่ได้รับ abiraterone acetate มากกว่าในผู้ป่วยที่ได้รับยาหลอก: hypokalaemia ตามลำดับ % เทียบกับ 11%, ความดันโลหิตสูง 16% เทียบกับ 11% และการกักเก็บของเหลว (บวมน้ำ) 26% เทียบกับ 20%. ในผู้ป่วยที่ได้รับการรักษาด้วย abiraterone acetate ภาวะ hypokalaemia ระดับ 3 และ 4 และความดันโลหิตสูงระดับ 3 และ 4 (Common Terminology Criteria for Adverse Events, CTCAE รุ่น 3.0) พบในผู้ป่วย 4% และ 2% ตามลำดับ ปฏิกิริยาของ mineralocorticoids ได้รับการจัดการทางเภสัชวิทยาด้วยผลลัพธ์ที่เป็นบวก การใช้คอร์ติโคสเตียรอยด์ร่วมกันช่วยลดอุบัติการณ์และความรุนแรงของอาการข้างเคียงเหล่านี้ (ดูหัวข้อ 4.4)
ตารางอาการไม่พึงประสงค์
การศึกษาที่ดำเนินการในผู้ป่วยมะเร็งต่อมลูกหมากระยะลุกลามขั้นสูง โดยได้รับ LHRH analogue หรือเคยได้รับ orchiectomy มาก่อน เกี่ยวข้องกับการให้ไซทีก้า 1,000 มก. ต่อวัน ร่วมกับยาเพรดนิโซนหรือเพรดนิโซโลนขนาดต่ำ (10 มก. ต่อ วัน).
อาการไม่พึงประสงค์จากยาที่สังเกตพบระหว่างการทดลองทางคลินิกและประสบการณ์หลังการขายจะระบุไว้ด้านล่างตามหมวดหมู่ความถี่ หมวดหมู่ความถี่มีการกำหนดดังนี้: ธรรมดามาก (≥ 1/10); ทั่วไป (≥ 1/100,
ภายในแต่ละประเภทความถี่จะรายงานผลกระทบที่ไม่พึงประสงค์ตามลำดับความรุนแรงที่ลดลง ตารางที่ 1: อาการไม่พึงประสงค์ที่ระบุในการศึกษาทางคลินิกและหลังการขาย
* ภาวะหัวใจล้มเหลวยังรวมถึงภาวะหัวใจล้มเหลว, หัวใจห้องล่างซ้ายล้มเหลว และส่วนที่ดีดออกลดลง
** กระดูกหักรวมถึงกระดูกหักทั้งหมดยกเว้นการแตกหักทางพยาธิวิทยา
รายงานที่เกิดขึ้นเองจากประสบการณ์หลังการขาย
ในผู้ป่วยที่ได้รับการรักษาด้วย abiraterone acetate อาการไม่พึงประสงค์ระดับ 3 ต่อไปนี้ (CTCAE เวอร์ชัน 3.0) เกิดขึ้น: hypokalaemia 3%; การติดเชื้อทางเดินปัสสาวะ, อะลานีนอะมิโนทรานสเฟอเรสเพิ่มขึ้น, ความดันโลหิตสูง, แอสพาเทตอะมิโนทรานสเฟอเรสเพิ่มขึ้น, กระดูกหัก 2%; อาการบวมน้ำที่ส่วนปลาย หัวใจล้มเหลว และภาวะหัวใจห้องบน 1% ต่อครั้ง ระดับ 3 hypertriglyceridaemia และ angina pectoris (CTCAE version 3.0) เกิดขึ้นใน
คำอธิบายของอาการไม่พึงประสงค์ที่เลือก
ปฏิกิริยาหัวใจและหลอดเลือด
การทดลองทางคลินิกทั้งระยะที่ 3 ไม่รวมผู้ป่วยที่มีความดันโลหิตสูงที่ไม่สามารถควบคุมได้ โรคหัวใจที่มีนัยสำคัญทางคลินิก หลักฐานจากกล้ามเนื้อหัวใจตาย หรือเหตุการณ์ atherothrombotic ภายใน 6 เดือนที่ผ่านมา โรคหลอดเลือดหัวใจตีบรุนแรงหรือไม่เสถียร หรือภาวะหัวใจล้มเหลว NYHA class III หรือ IV (การศึกษาที่ 301) o ภาวะหัวใจล้มเหลว class II - IV (การศึกษา 302) o การวัด apoplexy ของ cardiac ejection fraction apoplexy และการเสียชีวิตอย่างกะทันหันของหัวใจ ในการศึกษาทางคลินิกระยะที่ 3 อุบัติการณ์ของอาการไม่พึงประสงค์จากหลอดเลือดในผู้ป่วยที่ได้รับ abiraterone acetate เทียบกับผู้ป่วยที่ได้รับยาหลอก ได้แก่ ความดันโลหิตสูง 14.5% เทียบกับ 10.5%, ภาวะหัวใจห้องบน 3.4% เทียบกับ 3.4% อิศวร 2.8% เทียบกับ 1.7%, โรคหลอดเลือดหัวใจตีบ 1.9% เทียบกับ 0.9% ภาวะหัวใจล้มเหลว 1.9% เทียบกับ 0.6% และจังหวะ 1.1% เทียบกับ 0,4%.
พิษต่อตับ
ความเป็นพิษต่อตับที่มีระดับความสูงของ ALT, AST และบิลิรูบินทั้งหมดได้รับการรายงานในผู้ป่วยที่ได้รับการรักษาด้วย abiraterone acetateในการศึกษาทางคลินิกทั้งหมด มีรายงานผู้ป่วยประมาณ 4% ของผู้ป่วยที่ได้รับ abiraterone acetate ในผู้ป่วยที่ได้รับ abiraterone acetate ในระดับที่สูงขึ้น 3 เดือนแรกของการเริ่มต้นการรักษา ในการศึกษาทางคลินิก 301 ผู้ป่วยที่มีระดับพื้นฐาน ALT หรือ AST สูง มีแนวโน้มที่จะมีการทดสอบการทำงานของตับสูงกว่าผู้ป่วยที่เริ่มต้นด้วยค่าปกติ เมื่อเพิ่ม ALT หรือ AST> 5 x ULN หรือระดับบิลิรูบิน> ตรวจพบ ULN 3 x ยา abiraterone acetate ถูกยกเลิกหรือหยุด ในสองกรณี มีการทดสอบการทำงานของตับเพิ่มขึ้นอย่างเห็นได้ชัด (ดูหัวข้อ 4.4) ผู้ป่วย 2 รายที่มีการทำงานของตับปกติที่การตรวจวัดพื้นฐานมีระดับ ALT หรือ AST ตั้งแต่ 15 ถึง 40 x ULN และ ในบิลิรูบินตั้งแต่ 2 ถึง 6 x ULN การทดสอบการทำงานของตับในผู้ป่วยทั้งสองกลับสู่ภาวะปกติ และผู้ป่วยรายหนึ่งได้รับการรักษาซ้ำ โดยไม่เพิ่มค่าซ้ำแล้วซ้ำอีก ในการศึกษา 302 พบว่าระดับ 3 หรือ 4 ระดับความสูงใน ALT หรือ AST ในผู้ป่วย 35 ราย (6.5%) ที่ได้รับการรักษาด้วย abiraterone acetate ระดับของอะมิโนทรานส์เฟอเรสได้รับการแก้ไขในทั้งหมดยกเว้นผู้ป่วย 3 ราย (2 รายที่มีการแพร่กระจายของตับหลายครั้งและ 1 รายที่มีการยกระดับ AST ประมาณ 3 สัปดาห์หลังจากรับประทานยา abiraterone acetate ครั้งสุดท้าย) การหยุดชะงักของการรักษาเนื่องจากระดับ ALT และ AST มีรายงานในผู้ป่วย 1.7% และ 1.3% รักษาด้วย abiraterone acetate และ 0.2% และ 0% ของผู้ป่วยที่ได้รับยาหลอก ตามลำดับ ไม่มีรายงานการเสียชีวิตเนื่องจากเหตุการณ์ที่เป็นพิษต่อตับ
ในการทดลองทางคลินิก ความเสี่ยงของการเกิดพิษต่อตับลดลงโดยการยกเว้นผู้ป่วยที่เป็นโรคตับอักเสบที่การตรวจวัดพื้นฐานหรือมีความผิดปกติในการทดสอบการทำงานของตับอย่างมีนัยสำคัญ ในการทดลองทางคลินิก 301 ผู้ป่วยที่มีค่าพื้นฐาน ALT และ AST ≥ 2.5 x ULN โดยไม่มีการแพร่กระจายของตับและ> 5 x ULN ไม่รวมการแพร่กระจายของตับ ในการศึกษาทางคลินิก ผู้ป่วย 302 รายที่มีการแพร่กระจายของตับไม่มีสิทธิ์และไม่รวมผู้ป่วยที่เป็น baseline ALT และ AST ≥ 2.5 x ULN ความผิดปกติของการทดสอบการทำงานของตับที่พบในผู้ป่วยที่เข้าร่วมในการทดลองทางคลินิกได้รับการจัดการ แบบไดนามิกโดยหันไปใช้การหยุดชะงักของการรักษาและปล่อยให้การรักษาซ้ำหลังจากที่ค่าในการทดสอบการทำงานของตับกลับสู่ระดับพื้นฐานของผู้ป่วยแล้วเท่านั้น (ดูหัวข้อ 4.2) ผู้ป่วยที่มีระดับ ALT หรือ AST> 20 x ULN ไม่ได้รับการรักษาซ้ำ ไม่ทราบความปลอดภัยของการรักษาซ้ำในผู้ป่วยดังกล่าว ไม่ทราบกลไกของความเป็นพิษต่อตับที่เกี่ยวข้องกับไซทีก้า
การรายงานอาการไม่พึงประสงค์ที่น่าสงสัย
การรายงานอาการไม่พึงประสงค์ที่น่าสงสัยซึ่งเกิดขึ้นหลังจากการอนุมัติผลิตภัณฑ์ยามีความสำคัญเนื่องจากช่วยให้สามารถตรวจสอบความสมดุลของผลประโยชน์/ความเสี่ยงของผลิตภัณฑ์ยาได้อย่างต่อเนื่อง ขอให้ผู้เชี่ยวชาญด้านสุขภาพรายงานอาการไม่พึงประสงค์ที่น่าสงสัยผ่านระบบการรายงานระดับประเทศ "ที่อยู่ www. agenziafarmaco.gov.it/it/responsabili.
04.9 ยาเกินขนาด -
ประสบการณ์ของมนุษย์ในการใช้ยาเกินขนาดกับไซทีก้านั้นมีจำกัด
ไม่มียาแก้พิษเฉพาะ ในกรณีที่ให้ยาเกินขนาด ควรยุติการให้ยาและดำเนินมาตรการสนับสนุนทั่วไป รวมถึงการเฝ้าติดตามภาวะหัวใจเต้นผิดจังหวะ ภาวะโพแทสเซียมในเลือดต่ำ และอาการและอาการแสดงของการกักเก็บของเหลว ควรทำการประเมินการทำงานของตับด้วย
05.0 คุณสมบัติทางเภสัชวิทยา -
05.1 "คุณสมบัติทางเภสัชพลศาสตร์ -
กลุ่มเภสัชบำบัด: การรักษาต่อมไร้ท่อ ฮอร์โมนคู่อริอื่นๆ และสารที่เกี่ยวข้อง
รหัส ATC: L02BX03
กลไกการออกฤทธิ์
Abiraterone acetate (ZYTIGA) ถูกแปลง ในร่างกาย ใน abiraterone ซึ่งเป็นตัวยับยั้งการสังเคราะห์แอนโดรเจน โดยเฉพาะอย่างยิ่ง abiraterone เลือกยับยั้งเอนไซม์17α-hydroxylase / C17,20-lyase (CYP17) เอนไซม์นี้แสดงออกตามปกติและจำเป็นสำหรับการสังเคราะห์ฮอร์โมนแอนโดรเจนในเนื้อเยื่อต่อมลูกหมากต่อมลูกหมากอัณฑะและต่อมลูกหมาก CYP17 กระตุ้นการเปลี่ยนแปลงของ pregnenolone และ progesterone เป็นสารตั้งต้นของฮอร์โมนเทสโทสเตอโรน DHEA และ androstenedione ตามลำดับ โดย 17α-hydroxylation และความแตกแยกของพันธะ C17,20 การยับยั้ง CYP17 ยังทำให้การผลิตแร่คอร์ติคอยด์เพิ่มขึ้นโดยต่อมหมวกไต (ดูหัวข้อ 4.4)
มะเร็งต่อมลูกหมากที่ไวต่อแอนโดรเจนตอบสนองต่อการรักษาโดยการลดระดับแอนโดรเจน การบำบัดด้วยการกีดกันแอนโดรเจน เช่น การรักษาด้วย LHRH analogues หรือ orchiectomy ช่วยลดการผลิตแอนโดรเจนในอัณฑะ โดยไม่มีผลกระทบต่อการผลิตฮอร์โมนแอนโดรเจนโดยต่อมหมวกไตหรือในเนื้องอก การรักษาด้วยไซทีก้าช่วยลดระดับฮอร์โมนเทสโทสเตอโรนในซีรัมให้อยู่ในระดับที่ตรวจไม่พบ (โดยใช้การทดสอบเชิงพาณิชย์) เมื่อให้ยาด้วย LHRH analogues (หรือหลังการตัดไหม)
ผลกระทบทางเภสัชพลศาสตร์
ไซทีก้าลดฮอร์โมนเทสโทสเตอโรนและฮอร์โมนแอนโดรเจนอื่นๆ ให้อยู่ในระดับที่ต่ำกว่าที่ได้จากการใช้แอนะล็อก LHRH หรือ orchiectomy เพียงอย่างเดียว ผลกระทบนี้เป็นผลมาจากการเลือกยับยั้งเอนไซม์ CYP17 ที่จำเป็นสำหรับการสังเคราะห์แอนโดรเจน PSA ทำหน้าที่เป็นตัวบ่งชี้ทางชีวภาพใน ผู้ป่วยมะเร็งต่อมลูกหมาก ในการศึกษาทางคลินิกระยะที่ 3 ซึ่งดำเนินการในผู้ป่วยที่ดำเนินไปหลังการให้เคมีบำบัด Taxane ครั้งก่อน ผู้ป่วย 38% ที่ได้รับการรักษาด้วย abiraterone acetate มีระดับ PSA ลดลงอย่างน้อย 50% จากระดับพื้นฐาน เทียบกับ 10% ของผู้ป่วยที่ได้รับ ยาหลอก
ประสิทธิภาพและความปลอดภัยทางคลินิก
ประสิทธิภาพได้รับการจัดตั้งขึ้นในการทดลองทางคลินิกแบบ multicentre แบบ multicentre แบบ multicentre แบบสุ่มตัวอย่าง 2 ระยะ (การศึกษาที่ 301 และ 302) ในผู้ป่วยมะเร็งต่อมลูกหมากที่ทนต่อการแพร่กระจายของมะเร็งในระยะแพร่กระจาย ศึกษา 302 รายที่ลงทะเบียนโดยไม่รับการรักษาด้วย docetaxel และศึกษาผู้ป่วยที่ลงทะเบียนเรียน 301 รายซึ่งก่อนหน้านี้ ได้รับ docetaxel ผู้ป่วยใช้ LHRH analogue หรือเคยได้รับ orchiectomy ในกลุ่มบำบัดสารออกฤทธิ์ ไซทีก้าได้รับยาในขนาด 1,000 มก. ต่อวัน ร่วมกับยาเพรดนิโซนหรือเพรดนิโซโลนขนาดต่ำ 5 มก. วันละสองครั้ง ผู้ป่วยในกลุ่มควบคุมได้รับยาหลอกและยา prednisone หรือ prednisolone ขนาดต่ำ 5 มก. วันละสองครั้ง
ความผันแปรที่พบในความเข้มข้นของ PSA ในซีรัมแยกจากกันไม่ได้คาดการณ์ถึงประโยชน์ทางคลินิกเสมอไป ดังนั้น ในการทดลองทางคลินิกทั้งสองครั้ง ขอแนะนำให้รักษาผู้ป่วยตามแผนการรักษาด้วยการรักษาในการศึกษาที่ได้รับมอบหมาย จนกว่าจะตรงตามเกณฑ์การหยุดตามที่สรุปไว้ด้านล่างสำหรับการศึกษาทางคลินิกแต่ละครั้ง
ในการศึกษาทั้งสอง ไม่อนุญาตให้ใช้ spironolactone เพราะมันจับตัวรับแอนโดรเจนและสามารถเพิ่มระดับ PSA ได้
การศึกษา 302 (ผู้ป่วยเคมีบำบัดที่ไร้เดียงสา)
การศึกษานี้ลงทะเบียนผู้ป่วยเคมีบำบัดที่ไร้เดียงสาที่ไม่มีอาการหรือแสดงอาการเล็กน้อย และยังไม่ได้ระบุทางคลินิก อาการปวดรุนแรงขึ้นในช่วง 24 ชั่วโมงที่ผ่านมาด้วยคะแนน 0-1 ถือเป็นครั้งที่สองที่ไม่มีอาการ Brief Pain Inventory-Short Form (BPI-SF) และคะแนน 2-3 ถือว่าแสดงอาการเพียงเล็กน้อย
ในการศึกษา 302 (n = 1088) อายุมัธยฐานของผู้ป่วยที่ลงทะเบียนคือ 71 ปีสำหรับผู้ป่วยที่ได้รับ ZYTIGA plus prednisone หรือ prednisolone และ 70 ปีสำหรับผู้ป่วยที่ได้รับยาหลอกร่วมกับ prednisone หรือ prednisolone จำนวนผู้ป่วยที่รับการรักษาด้วย ZYTIGA ตามเชื้อชาติ กลุ่มคือ 520 คนผิวขาว (95.4%) 15 คนผิวดำ (2.8%) ชาวเอเชีย 4 คน (0.7%) และอีก 6 คน (1.1%)กลุ่มเนื้องอกวิทยาสหกรณ์ตะวันออก (ECOG) เป็น 0 สำหรับ 76% ของผู้ป่วยและ 1 สำหรับ 24% ของผู้ป่วยในแขนทั้งสองข้าง ผู้ป่วยห้าสิบเปอร์เซ็นต์มีการแพร่กระจายของกระดูกเท่านั้น อีก 31% ของผู้ป่วยมีการแพร่กระจายของกระดูกและเนื้อเยื่ออ่อนหรือต่อมน้ำเหลือง และ 19% ของผู้ป่วยมีเพียงเนื้อเยื่ออ่อนหรือการแพร่กระจายของต่อมน้ำเหลือง ไม่รวมผู้ป่วยที่มีการแพร่กระจายของอวัยวะภายใน NS ปลายทาง คะแนนประสิทธิภาพเบื้องต้นคือการรอดชีวิตโดยรวมและการรอดชีวิตที่ปราศจากความก้าวหน้าทางรังสี (rPFS) นอกเหนือจากขนาดของ ปลายทาง ประโยชน์ยังได้รับการประเมินโดยใช้เวลาในการใช้ฝิ่นสำหรับความเจ็บปวดจากมะเร็ง เวลาที่เริ่มให้เคมีบำบัดที่เป็นพิษต่อเซลล์ เวลาในการถดถอยของคะแนน ECOG ≥ 1 จุด และเวลาต่อการพัฒนา PSA ตามเกณฑ์ของ คณะทำงานมะเร็งต่อมลูกหมาก-2 (PCWG2). การรักษาในการศึกษาหยุดลงในช่วงเวลาของความก้าวหน้าทางคลินิกที่ชัดเจน นอกจากนี้ การรักษายังสามารถหยุดได้โดยขึ้นอยู่กับดุลยพินิจของผู้วิจัย ณ เวลาที่ยืนยันการลุกลามของรังสี
การรอดชีวิตที่ปราศจากความก้าวหน้าทางรังสี (rPFS) ได้รับการประเมินโดยใช้ การถ่ายภาพ ตามลำดับตามที่กำหนดโดยเกณฑ์ PCWG2 (สำหรับรอยโรคของกระดูก) และเกณฑ์ที่แก้ไขของ เกณฑ์การประเมินการตอบสนองในเนื้องอกที่เป็นของแข็ง (RECIST) (สำหรับการบาดเจ็บของเนื้อเยื่ออ่อน). การวิเคราะห์ rPFS ใช้การประเมินความก้าวหน้าทางรังสีที่ตรวจสอบจากส่วนกลาง
ที่ "การวิเคราะห์ตามกำหนดเวลาของ rPFS c" มีเหตุการณ์ 401 เหตุการณ์ 150 (28%) ของผู้ป่วยที่ได้รับ ZYTIGA และ 251 (46%) ของผู้ป่วยที่ได้รับยาหลอกมีหลักฐานทางรังสีของความก้าวหน้าหรือเสียชีวิต พบความแตกต่างอย่างมีนัยสำคัญใน rPFS ระหว่างกลุ่มการรักษา (ดูตารางที่ 2)
NE = ไม่ประมาณการ
* ค่า P ตามการทดสอบอันดับบันทึกที่ปรับสำหรับปัจจัยการแบ่งชั้น ECOG (0 หรือ 1)
** อัตราส่วนความเป็นอันตราย
อย่างไรก็ตาม การรวบรวมข้อมูลผู้ป่วยยังคงดำเนินต่อไปจนถึงวันที่วิเคราะห์ครั้งที่สอง โฆษณาชั่วคราว การอยู่รอดโดยรวม (การอยู่รอดโดยรวม - ระบบปฏิบัติการ) การตรวจรังสี rPFS ของผู้วิจัยแสดงไว้ในตารางที่ 3
ผู้ป่วยหกร้อยเจ็ดรายมีความก้าวหน้าทางรังสีวิทยาหรือเสียชีวิต: 271 (50%) ในกลุ่ม abiraterone acetate และ 336 (62%) ในกลุ่มยาหลอก การรักษาด้วย abiraterone acetate ลดความเสี่ยงของความก้าวหน้าทางรังสีวิทยาหรือการเสียชีวิตได้ 47% เมื่อเทียบกับยาหลอก (HR = 0.530; 95% CI: [0.451; 0.623], p
* ค่า P ตามการทดสอบอันดับบันทึกที่ปรับสำหรับปัจจัยการแบ่งชั้น ECOG (0 หรือ 1)
** อัตราส่วนความเป็นอันตราย
การวิเคราะห์ระหว่างกาลตามแผน (IA) สำหรับ OS ได้ดำเนินการหลังจากการสังเกตการเสียชีวิต 333 ราย จากผลการรักษาทางคลินิกที่มีนัยสำคัญที่สังเกตพบ การศึกษาได้เปิดขึ้น และให้การรักษาด้วย ZYTIGA แก่ผู้ป่วยในกลุ่มยาหลอก ลดความเสี่ยงของการเสียชีวิตลง 25% (HR = 0.752; 95% CI: [0.606; 0.934], p = 0.0097) แต่ OS ยังไม่สมบูรณ์และผลลัพธ์ในช่วงระยะเวลาหนึ่งไม่เป็นไปตามขีดจำกัดการหยุดเป้าหมายสำหรับนัยสำคัญทางสถิติ (ดู ตารางที่ 4). การอยู่รอดดำเนินต่อไปหลังจาก AI นี้
การวิเคราะห์ระบบปฏิบัติการตามแผนขั้นสุดท้ายดำเนินการหลังจากการสังเกตการเสียชีวิต 741 ราย (ติดตามค่ามัธยฐาน 49 เดือน) ผู้ป่วยร้อยละ 65 (354 จาก 546) ที่ได้รับการรักษาด้วย ZYTIGA เทียบกับ 71% (387 จาก 542) ของผู้ป่วย เสียชีวิตจากการรักษาด้วยยาหลอก ความได้เปรียบที่มีนัยสำคัญทางสถิติใน OS ในกลุ่ม ZYTIGA แสดงให้เห็นด้วยการลดความเสี่ยงต่อการเสียชีวิตลง 19.4% (HR = 0.806; 95% CI: [0.697; 0.931] , p = 0.0033) และ การปรับปรุงระบบปฏิบัติการเฉลี่ย 4.4 เดือน (ไซทีก้า 34.7 เดือน, ยาหลอก 30.3 เดือน) (ดูตารางที่ 4) การปรับปรุงนี้แสดงให้เห็นแม้ว่าผู้ป่วย 44% ที่ได้รับยาหลอกจะได้รับ ZYTIGA ในการรักษาภายหลัง
NE = ไม่ประมาณการ
* ค่า P ตามการทดสอบอันดับบันทึกที่ปรับสำหรับปัจจัยการแบ่งชั้น ECOG (0 หรือ 1)
** อัตราส่วนอันตราย
นอกจากการปรับปรุงที่สังเกตได้ในการรอดชีวิตโดยรวมและ rPFS แล้ว ยังแสดงให้เห็นประโยชน์ของการรักษาด้วยไซทีก้า เทียบกับ ยาหลอกทั้งหมด ปลายทาง รองดังนี้
ระยะเวลาในการลุกลามของ PSA ตามเกณฑ์ PCWG2: เวลามัธยฐานในการลุกลามของ PSA คือ 11.1 เดือนสำหรับผู้ป่วยที่ได้รับ ZYTIGA และ 5.6 เดือนสำหรับผู้ป่วยที่ได้รับยาหลอก (HR = 0.488; 95% CI : [0.420; 0.568], p
เวลาที่ต้องใช้ opioid สำหรับความเจ็บปวดจากมะเร็ง: เวลามัธยฐานของการใช้ยา opioid สำหรับความเจ็บปวดที่เกิดจากมะเร็งต่อมลูกหมาก ณ เวลาที่ทำการวิเคราะห์ขั้นสุดท้ายคือ 33.4 เดือนสำหรับผู้ป่วยที่ได้รับ ZYTIGA และ 23, 4 เดือนสำหรับผู้ป่วยที่ได้รับยาหลอก (HR = 0.721; 95% CI: [0.614, 0.846], p
เวลาในการให้เคมีบำบัดที่เป็นพิษต่อเซลล์: เวลามัธยฐานของเคมีบำบัดที่เป็นพิษต่อเซลล์คือ 25.2 เดือนสำหรับผู้ป่วยที่ได้รับ ZYTIGA และ 16.8 เดือนสำหรับผู้ป่วยที่ได้รับยาหลอก (HR = 0.580; 95% CI: [0.487; 0.691], p
เวลาที่คะแนน ECOG แย่ลง ≥ 1 คะแนน: เวลามัธยฐานของคะแนน ECOG ที่แย่ลง ≥ 1 คะแนน คือ 12.3 เดือนสำหรับผู้ป่วยที่ได้รับ ZYTIGA และ 10.9 เดือนสำหรับผู้ป่วยที่ได้รับยาหลอก (HR = 0.821; 95% CI: [0.714, 0.943], p = 0.0053)
จุดยุติต่อไปนี้แสดงให้เห็นถึงข้อได้เปรียบที่มีนัยสำคัญทางสถิติในการสนับสนุนการรักษาด้วยไซทีก้า:
การตอบสนองตามวัตถุประสงค์: การตอบสนองตามวัตถุประสงค์ถูกกำหนดเป็นเปอร์เซ็นต์ของผู้ป่วยที่มีโรคที่วัดได้ซึ่งบรรลุการตอบสนองทั้งหมดหรือบางส่วนตามเกณฑ์ RECIST (ขนาดต่อมน้ำเหลืองที่เส้นพื้นฐาน ≥ 2 ซม. ถือเป็นรอยโรคเป้าหมาย) เปอร์เซ็นต์ของผู้ป่วยที่เป็นโรคที่วัดได้ที่การตรวจวัดพื้นฐานโดยมีการตอบสนองตามวัตถุประสงค์คือ 36% ในกลุ่ม ZYTIGA และ 16% ในกลุ่มยาหลอก (p
ความเจ็บปวด: การรักษาด้วย ZYTIGA ช่วยลดความเสี่ยงต่อการลุกลามของความรุนแรงของความเจ็บปวดได้ 18% เมื่อเทียบกับกลุ่มที่ได้รับยาหลอก (p = 0.0490) ระยะเวลาเฉลี่ยในการลุกลามคือ 26.7 เดือนในกลุ่ม ZYTIGA และ 18, 4 เดือนในกลุ่มยาหลอก
เวลาที่ทำให้ FACT-P แย่ลง (คะแนนรวม): การรักษาด้วย ZYTIGA ลดความเสี่ยงที่ FACT-P (คะแนนรวม) แย่ลง 22% เมื่อเทียบกับยาหลอก (p = 0.0028) เวลามัธยฐานที่ทำให้ FACT-P แย่ลง (คะแนนรวม) คือ 12.7 เดือนในกลุ่ม ZYTIGA และ 8.3 เดือนในกลุ่มยาหลอก
การศึกษา 301 (ผู้ป่วยที่ได้รับเคมีบำบัดก่อนหน้านี้)
ศึกษาผู้ป่วยที่ลงทะเบียน 301 รายที่เคยได้รับ docetaxel ผู้ป่วยไม่จำเป็นต้องมีความคืบหน้าในระหว่าง docetaxel เนื่องจากความเป็นพิษต่อเคมีบำบัดนี้อาจนำไปสู่การหยุดยาได้ ผู้ป่วยยังคงศึกษาการรักษาจนถึงการลุกลามของ PSA (ได้รับการยืนยันว่าเพิ่มขึ้น 25% จากระดับพื้นฐานของผู้ป่วย / ระดับล่าง) พร้อมกับความก้าวหน้าทางรังสีวิทยาที่กำหนดโดยโปรโตคอล และความก้าวหน้าตามอาการหรือทางคลินิก ผู้ป่วยที่ได้รับการรักษาด้วยคีโตโคนาโซลก่อนหน้าสำหรับมะเร็งต่อมลูกหมากไม่รวมอยู่ในการศึกษานี้ แอล"ปลายทาง ประสิทธิภาพเบื้องต้นคือการอยู่รอดโดยรวม
อายุเฉลี่ยของผู้ป่วยที่ลงทะเบียนคือ 69 ปี (ช่วง 39-95) จำนวนผู้ป่วยที่รักษาด้วย ZYTIGA โดยกลุ่มเชื้อชาติคือ 737 คนผิวขาว (93.2%), 28 คนผิวดำ (3.5%), 11 คนเอเชีย (1.4%) และอีก 14 คน (1.8%) 11% ของผู้ป่วยที่ลงทะเบียนเรียนมีคะแนน คะแนนประสิทธิภาพ ตามมาตราส่วน ECOG 2; 70% นำเสนอหลักฐานการถ่ายภาพรังสีของการลุกลามของโรคโดยมีหรือไม่มีความก้าวหน้าของ PSA 70% เคยได้รับเคมีบำบัดที่เป็นพิษต่อเซลล์ก่อนหน้านี้ และ 30% มีสองครั้ง มีการแพร่กระจายของตับในผู้ป่วย 11% ที่ได้รับ ZYTIGA
ในการวิเคราะห์ตามแผนซึ่งดำเนินการหลังจากเสียชีวิต 552 ราย ผู้ป่วย 42% (333 จาก 797) ที่ได้รับการรักษาด้วย ZYTIGA เสียชีวิต เทียบกับ 55% (219 จาก 398) ของผู้ป่วยที่ได้รับยาหลอก พบว่ายาหลอกดีขึ้นอย่างมีนัยสำคัญทางสถิติ . ค่ามัธยฐานการรอดชีวิตโดยรวมของผู้ป่วยที่รักษาด้วยไซทีก้า (ดูตารางที่ 5)
ค่า p ตามการทดสอบอันดับบันทึกที่ปรับสำหรับปัจจัยการแบ่งชั้น ECOG (0-1 เทียบกับ 2) คะแนนความเจ็บปวด (ไม่มีเทียบกับปัจจุบัน) จำนวนสูตรเคมีบำบัดก่อนหน้า (1 ต่อ 2) และประเภทของความก้าวหน้าของโรค (เท่านั้น PSA กับรังสี)
b อัตราส่วนความเป็นอันตรายตามแบบจำลองความเสี่ยงที่ปรับตามปัจจัยการแบ่งชั้น อัตราส่วนความเสี่ยง
ในการประเมินทุกขั้นตอน หลังจากการรักษาในช่วง 2-3 เดือนแรก สัดส่วนของผู้ป่วยที่รักษาด้วย ZYTIGA ยังคงมีชีวิตสูงกว่าสัดส่วนของผู้ป่วยที่ได้รับยาหลอก
การวิเคราะห์กลุ่มย่อยการรอดชีวิตแสดงให้เห็นประโยชน์ในการรอดชีวิตอย่างมีนัยสำคัญสำหรับการรักษาด้วยไซทีก้า
นอกเหนือจากการปรับปรุงที่สังเกตได้ในการอยู่รอดโดยรวมแล้ว ทั้งหมด ปลายทาง การศึกษาระดับมัธยมศึกษาเห็นด้วยกับไซทีก้า เช่นเดียวกับการมีนัยสำคัญทางสถิติหลังจากปรับสำหรับการทดลองหลายครั้ง โดยอิงจากข้อมูลต่อไปนี้:
ผู้ป่วยที่รักษาด้วย ZYTIGA มีอัตราการตอบสนอง PSA โดยรวมที่สูงกว่าอย่างมีนัยสำคัญ (หมายถึงการลดลง ≥ 50% จากค่าพื้นฐาน) เมื่อเทียบกับผู้ป่วยที่ได้รับยาหลอก, 38% เทียบกับ 10%, p
เวลาเฉลี่ยในการลุกลามของ PSA คือ 10.2 เดือนสำหรับผู้ป่วยที่ได้รับ ZYTIGA และ 6.6 เดือนสำหรับผู้ป่วยที่ได้รับยาหลอก (HR = 0.580; 95% CI: [0.462; 0.728], p
การรอดชีวิตที่ปราศจากความก้าวหน้าเฉลี่ยตามที่กำหนดโดยการตรวจทางรังสีคือ 5.6 เดือนสำหรับผู้ป่วยที่ได้รับ ZYTIGA และ 3.6 เดือนสำหรับผู้ป่วยที่ได้รับยาหลอก (HR = 0.673; 95% CI: [0.585; 0.776 ], p
ปวด
เปอร์เซ็นต์ของผู้ป่วยที่รายงานการบรรเทาอาการปวดในกลุ่ม ZYTIGA มีนัยสำคัญทางสถิติมากกว่าในกลุ่มที่ได้รับยาหลอก (44% เทียบกับ 27%, p = 0.0002) ตอบกลับ สำหรับการบรรเทาอาการปวดถูกกำหนดให้เป็นผู้ป่วยที่มีประสบการณ์การลดลงอย่างน้อย 30% จากระดับพื้นฐานในคะแนนความรุนแรงของความเจ็บปวดที่เลวร้ายที่สุดตาม BPI SF ในช่วง 24 ชั่วโมงที่ผ่านมาโดยไม่มีคะแนนการใช้ยาแก้ปวดเพิ่มขึ้น สังเกตได้จากการประเมินสองครั้งติดต่อกัน ห่างกัน 4 สัปดาห์ วิเคราะห์เฉพาะผู้ป่วยที่มีคะแนน ≥ 4 และคะแนนความเจ็บปวดหลังการตรวจวัดพื้นฐานอย่างน้อยหนึ่งคะแนน (N = 512) เพื่อบรรเทาอาการปวด
เปอร์เซ็นต์ของผู้ป่วยที่รักษาด้วย ZYTIGA มีความเจ็บปวดน้อยกว่าผู้ป่วยที่ได้รับยาหลอกที่ 6 (22% vs 28%), 12 (30% vs 38%) และ 18 เดือน (35% vs 46%) ความก้าวหน้าของความเจ็บปวดถูกกำหนดให้เพิ่มขึ้น ≥ 30% จากค่าพื้นฐานในคะแนนความรุนแรงของความเจ็บปวดที่แย่ที่สุดของ BPI SF ในช่วง 24 ชั่วโมงก่อนหน้า โดยไม่มีการลดลงในคะแนนการใช้ยาแก้ปวดที่สังเกตพบในการนัดตรวจสองครั้งติดต่อกัน หรือคะแนนการใช้ยาแก้ปวดเพิ่มขึ้น ≥ 30% ที่สังเกตพบ การเข้ารับการตรวจติดต่อกัน 2 ครั้ง ระยะเวลาในการลุกลามของความเจ็บปวดไปถึงเปอร์เซ็นไทล์ที่ 25 คือ 7.4 เดือนในกลุ่ม ZYTIGA เทียบกับ 4.7 เดือนของกลุ่มยาหลอก
เหตุการณ์ที่ส่งผลต่อระบบโครงกระดูก
เปอร์เซ็นต์ที่ต่ำกว่าของผู้ป่วยในกลุ่ม ZYTIGA พบเหตุการณ์เกี่ยวกับระบบโครงร่าง เมื่อเทียบกับผู้ป่วยในกลุ่มยาหลอกที่ 6 เดือน (18% เทียบกับ 28%), 12 เดือน (30% vs 40%) และ 18 เดือน (35% vs 40%) . ในกลุ่มที่บำบัดด้วยไซทีก้า เวลาที่เกิดโครงกระดูกครั้งแรกที่เปอร์เซ็นไทล์ที่ 25 เป็นสองเท่าของกลุ่มควบคุมที่ 9.9 เดือน เทียบกับ 4.9 เดือน เหตุการณ์ของระบบโครงร่างหมายถึงการแตกหักทางพยาธิวิทยา การกดทับของไขสันหลัง การฉายรังสีแบบประคับประคองไปยังกระดูก หรือการผ่าตัดไปที่กระดูก
ประชากรเด็ก
European Medicines Agency ได้ยกเว้นภาระหน้าที่ในการส่งผลการศึกษากับ ZYTIGA ในกลุ่มย่อยทั้งหมดของประชากรเด็กที่เป็นมะเร็งต่อมลูกหมากขั้นสูง ดูหัวข้อ 4.2 สำหรับข้อมูลเกี่ยวกับการใช้ในเด็ก
05.2 "คุณสมบัติทางเภสัชจลนศาสตร์ -
หลังจากได้รับ abiraterone acetate แล้ว ข้อมูลทางเภสัชจลนศาสตร์ของ abiraterone และ abiraterone acetate ได้รับการศึกษาในคนที่มีสุขภาพดี ในผู้ป่วยมะเร็งระยะลุกลามขั้นสูงของต่อมลูกหมาก และในผู้ป่วยที่ไม่เป็นมะเร็งที่มีความบกพร่องทางตับหรือไต Abiraterone acetate ถูกแปลงอย่างรวดเร็ว ในร่างกาย ใน abiraterone ซึ่งเป็นตัวยับยั้งการสังเคราะห์แอนโดรเจน (ดูหัวข้อ 5.1)
การดูดซึม
หลังจากการให้ abiraterone acetate ทางปากในสภาวะอดอาหาร เวลาที่ต้องใช้เพื่อให้ได้ความเข้มข้นสูงสุดของ abiraterone ในพลาสมาจะอยู่ที่ประมาณ 2 ชั่วโมง
การบริหาร abiraterone acetate กับอาหาร เมื่อเทียบกับการให้ยาในสภาวะที่อดอาหาร ส่งผลให้มีการได้รับ abiraterone เฉลี่ยทั้งระบบเพิ่มขึ้นถึง 10 เท่า [AUC] และสูงขึ้นถึง 17 เท่า [Cmax] โดยพิจารณาจากไขมันที่มีอยู่ในอาหาร การรับประทานไซทีก้าร่วมกับอาหารอาจส่งผลให้ได้รับสารที่เปลี่ยนแปลงได้สูง ดังนั้นจึงไม่ควรรับประทานไซทีก้าร่วมกับอาหาร ควรรับประทานก่อนหรือหลังอาหารอย่างน้อย 1 ชั่วโมงหรืออย่างน้อย 2 ชั่วโมง ควรกลืนเม็ดยาทั้งเม็ดด้วยน้ำบางส่วน (ดูหัวข้อ 4.2)
การกระจาย
การจับกันของ abiraterone ที่ติดฉลาก 14C กับโปรตีนในพลาสมาคือ 99.8% ปริมาตรที่ชัดเจนของการกระจายอยู่ที่ประมาณ 5,630 ลิตร ซึ่งบ่งชี้ถึงการกระจายตัวของอะบิเรเทอโรนอย่างกว้างขวางในเนื้อเยื่อส่วนปลาย
การเปลี่ยนแปลงทางชีวภาพ
ภายหลังการให้ abiraterone acetate ที่ติดฉลากกัมมันตภาพรังสี 14C ในแคปซูล abiraterone acetate จะถูกไฮโดรไลซ์ไปเป็น abiraterone ซึ่งจะต้องผ่านกระบวนการเมแทบอลิซึม ซึ่งรวมถึงการเกิดซัลเฟต การเกิดไฮดรอกซิเลชัน และการออกซิเดชัน โดยเฉพาะอย่างยิ่งในตับ กัมมันตภาพรังสีส่วนใหญ่ที่มีอยู่ในการไหลเวียน (ประมาณ 92%) พบได้ในรูปของสารเมแทบอไลต์ของ abiraterone สารเมแทบอไลต์หลัก 2 ชนิดจากอะบิเรเทอโรนซัลเฟตที่ตรวจพบได้ 15 ชนิดและไนออกไซด์อะบิเรเทอโรนซัลเฟตซึ่งแต่ละชนิดมีสัดส่วนประมาณ 43% ของกัมมันตภาพรังสีทั้งหมด
การกำจัด
ครึ่งชีวิตเฉลี่ยของ abiraterone ในพลาสมาอยู่ที่ประมาณ 15 ชั่วโมง โดยอิงจากข้อมูลจากอาสาสมัครที่มีสุขภาพดี หลังจากได้รับ abiraterone acetate ที่ติดฉลากกัมมันตภาพรังสี 14C ขนาด 1,000 มก. พบว่าประมาณ 88% ของขนาดยากัมมันตภาพรังสีถูกกู้คืนในอุจจาระและ 5% circanell "ปัสสาวะ สารประกอบหลักที่มีอยู่ในอุจจาระคือ abiraterone acetate และ abiraterone ไม่เปลี่ยนแปลง (ประมาณ 55% และ 22% ของขนาดยาตามลำดับ)
การด้อยค่าของตับ
เภสัชจลนศาสตร์ของ abiraterone acetate ได้รับการตรวจสอบในอาสาสมัครที่มีความบกพร่องของตับในระดับเล็กน้อยหรือปานกลางที่มีอยู่ก่อนแล้ว (Child-Pugh Class A และ B ตามลำดับ) และในกลุ่มควบคุมที่มีสุขภาพดี การได้รับ abiraterone อย่างเป็นระบบหลังจากรับประทานครั้งเดียวขนาด 1,000 มก. เพิ่มขึ้นประมาณ 11% และ 260% ตามลำดับในผู้ที่มีความบกพร่องของตับในระดับเล็กน้อยและปานกลางที่มีอยู่ก่อน ครึ่งชีวิตเฉลี่ยของ abiraterone ถูกยืดออกไปประมาณ 18 ชั่วโมงในผู้ที่มีความบกพร่องทางตับเล็กน้อยและประมาณ 19 ชั่วโมงในผู้ที่มีตับบกพร่องในระดับปานกลาง
ในการศึกษาทางคลินิกอื่น เภสัชจลนศาสตร์ของ abiraterone ได้รับการตรวจสอบในอาสาสมัครที่มีความบกพร่องของตับอย่างรุนแรงที่มีอยู่ก่อน (n = 8) (Child-Pugh Class C) และใน 8 คนที่ได้รับการควบคุมที่มีสุขภาพดีที่มีการทำงานของตับตามปกติ AUC ของ abiraterone เพิ่มขึ้นประมาณ 600% และเศษของยาฟรี 80% ในผู้ป่วยที่มีความบกพร่องทางตับอย่างรุนแรงเมื่อเทียบกับผู้ที่มีการทำงานของตับปกติ
ไม่จำเป็นต้องปรับขนาดยาสำหรับผู้ป่วยที่มีภาวะตับบกพร่องเพียงเล็กน้อย
การใช้ abiraterone acetate ควรพิจารณาด้วยความระมัดระวังในผู้ป่วยที่มีความบกพร่องทางตับในระดับปานกลางซึ่งผลประโยชน์ต้องมีมากกว่าความเสี่ยงที่เป็นไปได้อย่างชัดเจน (ดูหัวข้อ 4.2 และ 4.4) ไม่ควรใช้ Abiraterone acetate ในผู้ป่วยที่มีความบกพร่องทางตับอย่างรุนแรง (ดูหัวข้อ 4.2 , 4.3 และ 4.4)
สำหรับผู้ป่วยที่เป็นพิษต่อตับระหว่างการรักษา อาจจำเป็นต้องระงับการรักษาและปรับขนาดยา (ดูหัวข้อ 4.2 และ 4.4).
การด้อยค่าของไต
เภสัชจลนศาสตร์ของ abiraterone acetate ถูกเปรียบเทียบในผู้ป่วยที่เป็นโรคไตวายเรื้อรังระยะสุดท้ายซึ่งมีตารางการฟอกไตที่เสถียรกับกลุ่มควบคุมที่เข้าคู่กับการทำงานของไตตามปกติ การได้รับ abiraterone อย่างเป็นระบบหลังจากรับประทานยาขนาด 1000 มก. ครั้งเดียวไม่เพิ่มขึ้นในผู้ป่วยที่เป็นโรคไตวายเรื้อรังระยะสุดท้ายที่ได้รับการฟอกไต การให้ยาแก่ผู้ป่วยไตวายเรื้อรังรวมถึงอาการรุนแรงไม่จำเป็นต้องลดขนาดยา (ดูหัวข้อ 4.2 อย่างไรก็ตาม ไม่มีประสบการณ์ทางคลินิก ในผู้ป่วยมะเร็งต่อมลูกหมากและการด้อยค่าของไตอย่างรุนแรง ผู้ป่วยเหล่านี้ควรระมัดระวัง
05.3 ข้อมูลความปลอดภัยพรีคลินิก -
ในการศึกษาความเป็นพิษต่อสัตว์ทั้งหมด พบว่าระดับฮอร์โมนเทสโทสเตอโรนหมุนเวียนลดลงอย่างมีนัยสำคัญ เป็นผลให้น้ำหนักอวัยวะลดลงและการเปลี่ยนแปลงทางสัณฐานวิทยาและ / หรือทางจุลพยาธิวิทยาในอวัยวะสืบพันธุ์และต่อมหมวกไต, ต่อมใต้สมองและเต้านม การเปลี่ยนแปลงทั้งหมดแสดงให้เห็นการย้อนกลับทั้งหมดหรือบางส่วน การเปลี่ยนแปลงของอวัยวะสืบพันธุ์และความไวต่อฮอร์โมนแอนโดรเจนนั้นเข้ากันได้กับเภสัชวิทยาของ abiraterone การเปลี่ยนแปลงของฮอร์โมนที่เกี่ยวข้องกับยาทั้งหมดกลับรายการหรือแก้ไขได้หลังจากช่วงพักฟื้น 4 สัปดาห์
ในการศึกษาภาวะเจริญพันธุ์ในหนูทั้งตัวผู้และตัวเมีย abiraterone acetate ช่วยลดภาวะเจริญพันธุ์ ซึ่งเป็นผลที่สามารถย้อนกลับได้เต็มที่ 4 ถึง 16 สัปดาห์หลังจากหยุดใช้ abiraterone acetate
ในการศึกษาความเป็นพิษต่อพัฒนาการในหนู abiraterone acetate ส่งผลต่อการตั้งครรภ์ รวมทั้งน้ำหนักของทารกในครรภ์ที่ลดลงและการรอดชีวิต สังเกตผลกระทบต่ออวัยวะเพศภายนอกแม้ว่า abiraterone acetate จะไม่ก่อให้เกิดการก่อมะเร็ง
ในการศึกษาภาวะเจริญพันธุ์และความเป็นพิษต่อพัฒนาการของหนู ผลกระทบทั้งหมดมีความสัมพันธ์กับกิจกรรมทางเภสัชวิทยาของ abiraterone acetate
นอกเหนือจากความผันแปรที่พบในอวัยวะสืบพันธุ์ในการศึกษาทางพิษวิทยาทั้งหมดที่ดำเนินการในสัตว์แล้ว ข้อมูลที่ไม่ใช่ทางคลินิกเผยให้เห็นว่าไม่มีอันตรายเป็นพิเศษสำหรับมนุษย์จากการศึกษาทั่วไปของ เภสัชวิทยาความปลอดภัย, ความเป็นพิษเมื่อให้ยาซ้ำ ความเป็นพิษต่อพันธุกรรม และศักยภาพในการก่อมะเร็ง Abiraterone acetate ไม่ก่อมะเร็งในการศึกษา 6 เดือนในหนูดัดแปลงพันธุกรรม (Tg.rasH2) ในการศึกษาสารก่อมะเร็งในหนูทดลอง 24 เดือน abiraterone acetate เพิ่มอุบัติการณ์ของเนื้องอกในเซลล์คั่นระหว่างหน้าในอัณฑะ การค้นพบนี้เชื่อว่าเกี่ยวข้องกับการกระทำทางเภสัชวิทยาของ abiraterone และมีความเฉพาะเจาะจงในหนู Abiraterone acetate ไม่ก่อมะเร็งในหนูเพศเมีย
สารออกฤทธิ์ abiraterone มีความเสี่ยงต่อสิ่งแวดล้อมในน้ำ โดยเฉพาะกับปลา
06.0 ข้อมูลทางเภสัชกรรม -
06.1 สารเพิ่มปริมาณ -
ไมโครคริสตัลลีน เซลลูโลส
ครอสคาร์เมลโลสโซเดียม
แลคโตสโมโนไฮเดรต
แมกนีเซียมสเตียเรต
โพวิโดน (K29 / K32)
ปราศจากน้ำคอลลอยด์ซิลิกา
โซเดียมลอริลซัลเฟต
06.2 ความเข้ากันไม่ได้ "-
ไม่เกี่ยวข้อง
06.3 ระยะเวลาที่มีผลใช้บังคับ "-
2 ปี.
06.4 ข้อควรระวังพิเศษสำหรับการจัดเก็บ -
ยานี้ไม่ต้องการเงื่อนไขการจัดเก็บพิเศษใด ๆ
06.5 ลักษณะการบรรจุทันทีและเนื้อหาของบรรจุภัณฑ์ -
ขวดพลาสติกโพลีเอทิลีนความหนาแน่นสูงทรงกลมสีขาว ปิดด้วยโพลีโพรพิลีนทนเด็ก บรรจุ 120 เม็ด แต่ละแพ็คมีหนึ่งขวด
06.6 คำแนะนำสำหรับการใช้งานและการจัดการ -
เนื่องจากกลไกการออกฤทธิ์ ยานี้อาจเป็นอันตรายต่อทารกในครรภ์ที่กำลังพัฒนา ดังนั้น สตรีมีครรภ์หรืออยู่ในวัยเจริญพันธุ์ไม่ควรรับประทานโดยไม่ใช้อุปกรณ์ป้องกัน เช่น ถุงมือ
ยาที่ไม่ได้ใช้และของเสียที่ได้จากยานี้ต้องกำจัดตามระเบียบข้อบังคับของท้องถิ่น ยานี้อาจก่อให้เกิดความเสี่ยงต่อสิ่งแวดล้อมในน้ำ (ดูหัวข้อ 5.3)
07.0 ผู้ถือ "การอนุญาตการตลาด" -
Janssen-Cilag International NV
Turnhoutseweg 30
B-2340 เบียร์เซ
เบลเยียม
08.0 หมายเลขอนุญาตการตลาด -
EU / 1/11/714/001
041427016
09.0 วันที่อนุญาตครั้งแรกหรือต่ออายุการอนุญาต -
วันที่ให้สิทธิ์ครั้งแรก: 05 กันยายน 2011
วันที่ต่ออายุครั้งล่าสุด: 26 พฤษภาคม 2016
10.0 วันที่แก้ไขข้อความ -
11/2016














.jpg)